

Oculocutaneous albinism
- PMID: 17980020
- PMCID: PMC2211462
- DOI: 10.1186/1750-1172-2-43
Oculocutaneous albinism
Abstract
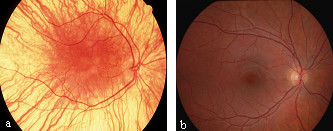
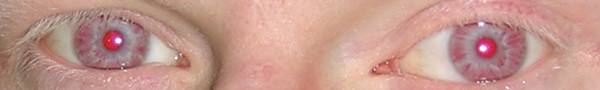
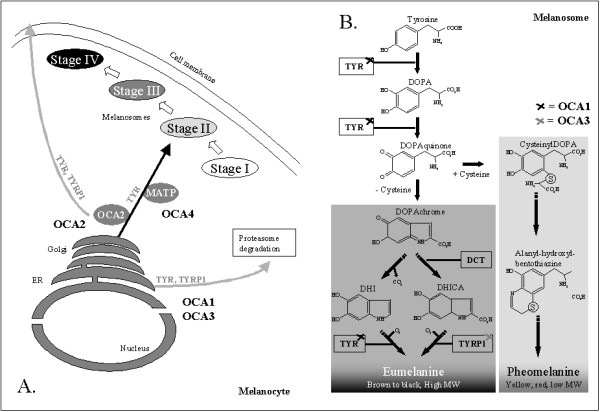
Oculocutaneous albinism (OCA) is a group of inherited disorders of melanin biosynthesis characterized by a generalized reduction in pigmentation of hair, skin and eyes. The prevalence of all forms of albinism varies considerably worldwide and has been estimated at approximately 1/17,000, suggesting that about 1 in 70 people carry a gene for OCA. The clinical spectrum of OCA ranges, with OCA1A being the most severe type with a complete lack of melanin production throughout life, while the milder forms OCA1B, OCA2, OCA3 and OCA4 show some pigment accumulation over time. Clinical manifestations include various degrees of congenital nystagmus, iris hypopigmentation and translucency, reduced pigmentation of the retinal pigment epithelium, foveal hypoplasia, reduced visual acuity usually (20/60 to 20/400) and refractive errors, color vision impairment and prominent photophobia. Misrouting of the optic nerves is a characteristic finding, resulting in strabismus and reduced stereoscopic vision. The degree of skin and hair hypopigmentation varies with the type of OCA. The incidence of skin cancer may be increased. All four types of OCA are inherited as autosomal recessive disorders. At least four genes are responsible for the different types of the disease (TYR, OCA2, TYRP1 and MATP). Diagnosis is based on clinical findings of hypopigmentation of the skin and hair, in addition to the characteristic ocular symptoms. Due to the clinical overlap between the OCA forms, molecular diagnosis is necessary to establish the gene defect and OCA subtype. Molecular genetic testing of TYR and OCA2 is available on a clinical basis, while, at present, analysis of TYRP1 and MATP is on research basis only. Differential diagnosis includes ocular albinism, Hermansky-Pudlak syndrome, Chediak-Higashi syndrome, Griscelli syndrome, and Waardenburg syndrome type II. Carrier detection and prenatal diagnosis are possible when the disease causing mutations have been identified in the family. Glasses (possibly bifocals) and dark glasses or photocromic lenses may offer sufficient help for reduced visual activity and photophobia. Correction of strabismus and nystagmus is necessary and sunscreens are recommended. Regular skin checks for early detection of skin cancer should be offered. Persons with OCA have normal lifespan, development, intelligence and fertility.
Figures
References
-
- Witkop CJ. Albinism: hematologic-storage disease, susceptibility to skin cancer, and optic neuronal defects shared in all types of oculocutaneous and ocular albinism. Ala J Med Sci. 1979;16:327–330. - PubMed
-
- Lee ST, Nicholls RD, Schnur RE, Guida LC, Lu-Kuo J, Spinner NB, Zackai EH, Spritz RA. Diverse mutations of the P gene among African-Americans with type II (tyrosinase-positive) oculocutaneous albinism (OCA2) Hum Mol Genet. 1994;3:2047–2051. - PubMed
-
- King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In: Scriver CR, Beaudet AL, Sly WS and Valle D, editor. The Metabolic and Molecular bases of inherited Disease. New York, McGraw-Hill, Inc.; 1995. pp. 4353–4392.
-
- Kromberg JG, Jenkins T. Prevalence of albinism in the South African negro. S Afr Med J. 1982;61:383–386. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
