

The genetic basis of a craniofacial disease provides insight into COPII coat assembly
- PMID: 17981132
- PMCID: PMC2262049
- DOI: 10.1016/j.devcel.2007.10.005
The genetic basis of a craniofacial disease provides insight into COPII coat assembly
Abstract
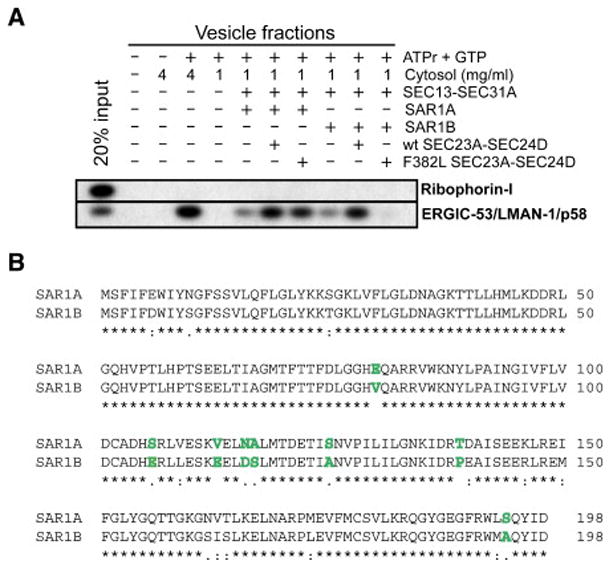
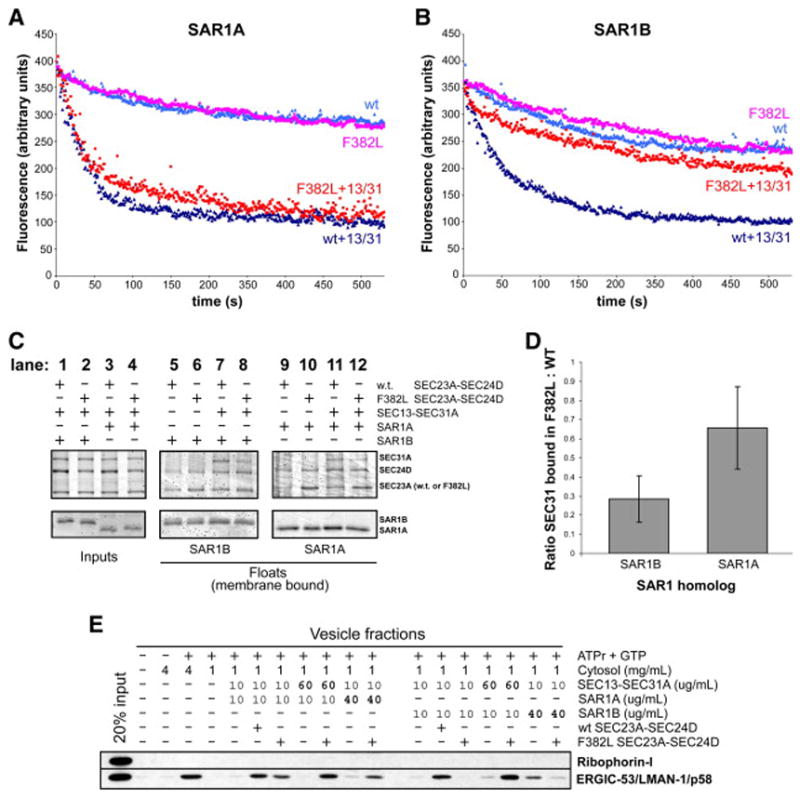
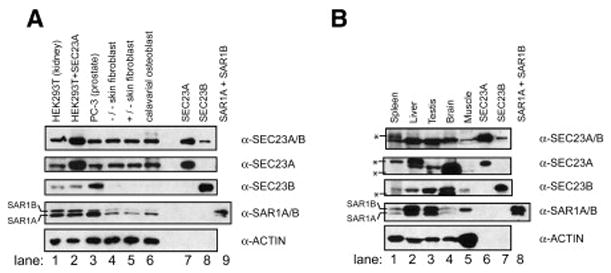
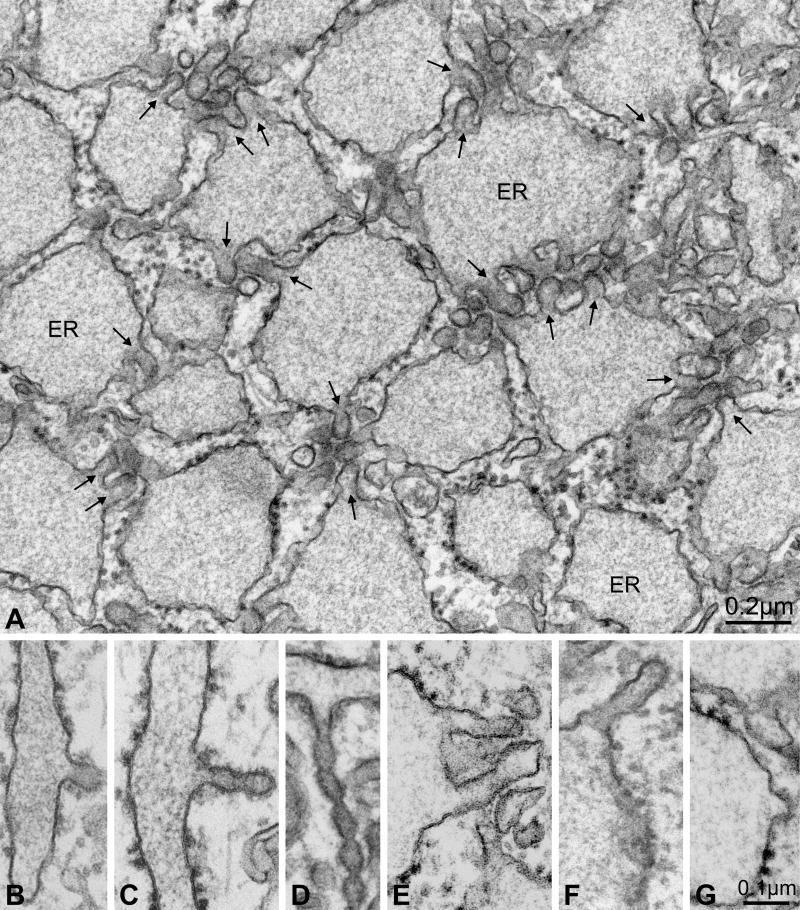
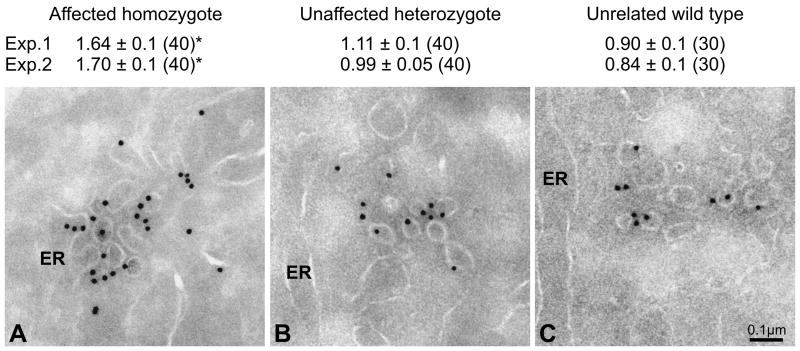
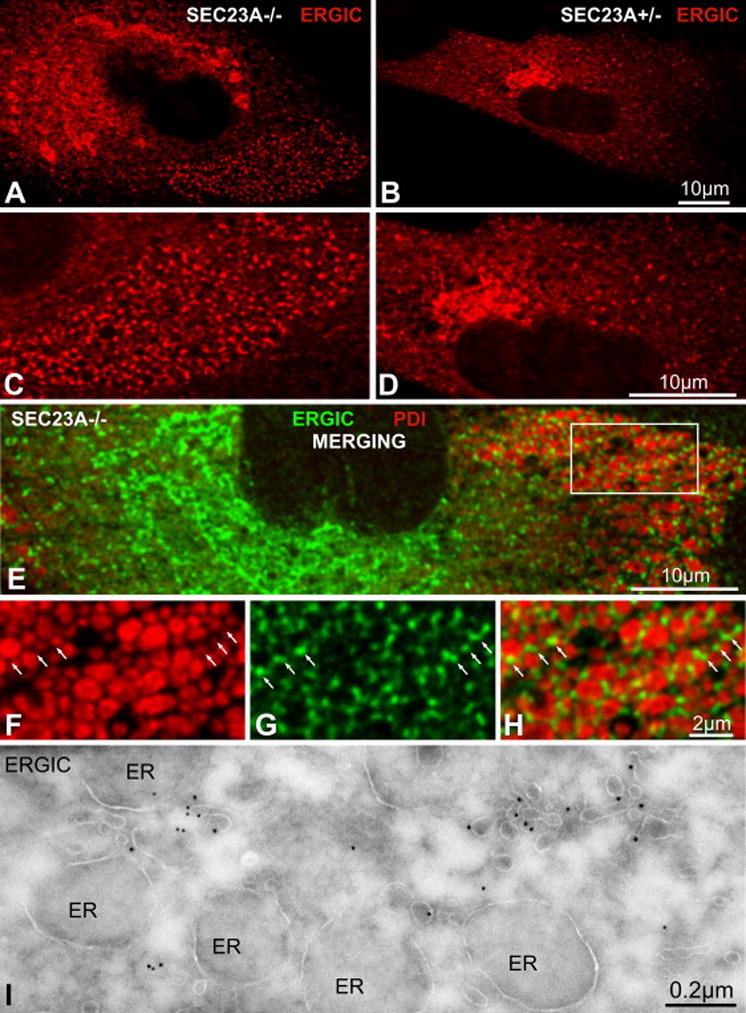
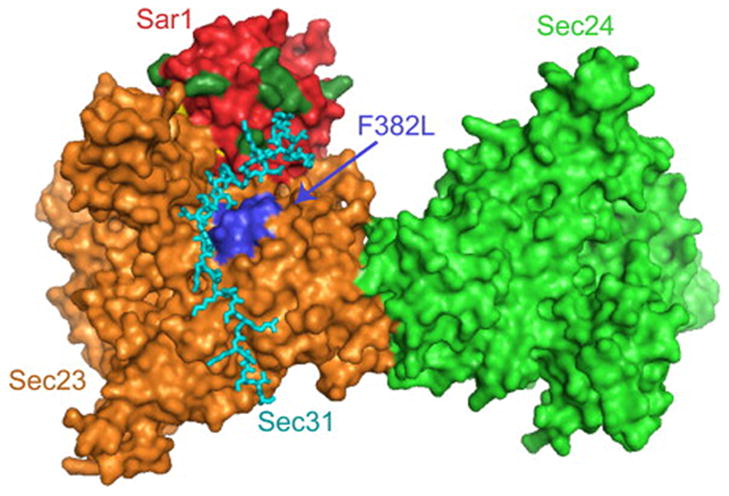
Proteins trafficking through the secretory pathway must first exit the endoplasmic reticulum (ER) through membrane vesicles created and regulated by the COPII coat protein complex. Cranio-lenticulo-sutural dysplasia (CLSD) was recently shown to be caused by a missense mutation in SEC23A, a gene encoding one of two paralogous COPII coat proteins. We now elucidate the molecular mechanism underlying this disease. In vitro assays reveal that the mutant form of SEC23A poorly recruits the Sec13-Sec31 complex, inhibiting vesicle formation. Surprisingly, this effect is modulated by the Sar1 GTPase paralog used in the reaction, indicating distinct affinities of the two human Sar1 paralogs for the Sec13-Sec31 complex. Patient cells accumulate numerous tubular cargo-containing ER exit sites devoid of observable membrane coat, likely representing an intermediate step in COPII vesicle formation. Our results indicate that the Sar1-Sec23-Sec24 prebudding complex is sufficient to form cargo-containing tubules in vivo, whereas the Sec13-Sec31 complex is required for membrane fission.
Figures
References
-
- Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–537. - PubMed
-
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. - PubMed
-
- Barlowe C, Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
