

Characterization of the resting MscS: modeling and analysis of the closed bacterial mechanosensitive channel of small conductance
- PMID: 17981908
- PMCID: PMC2212706
- DOI: 10.1529/biophysj.107.110171
Characterization of the resting MscS: modeling and analysis of the closed bacterial mechanosensitive channel of small conductance
Abstract
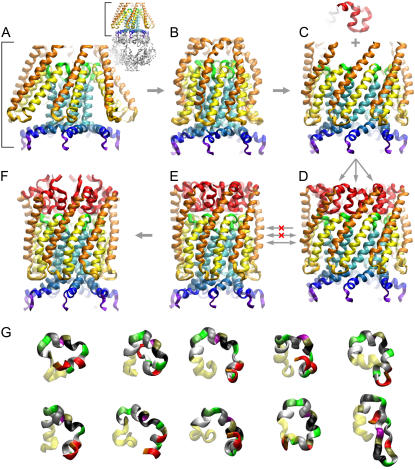
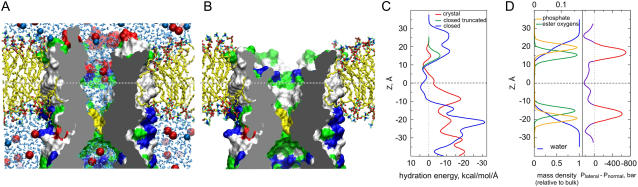
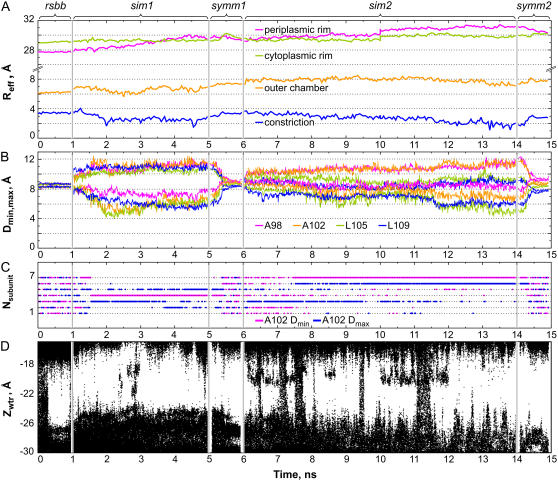
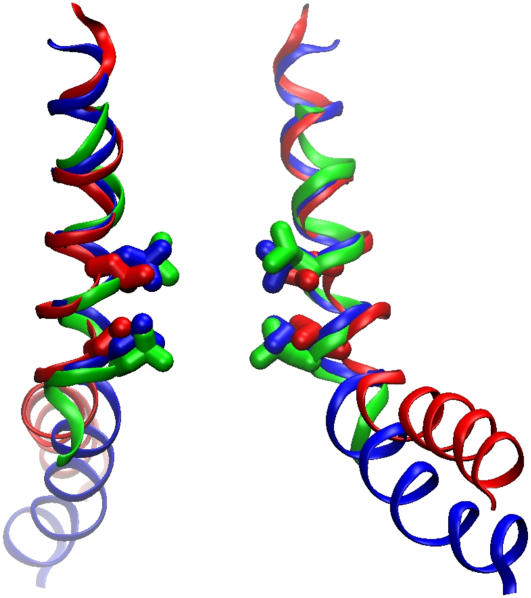
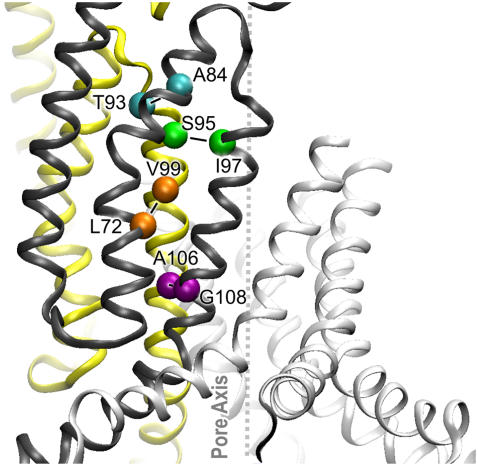
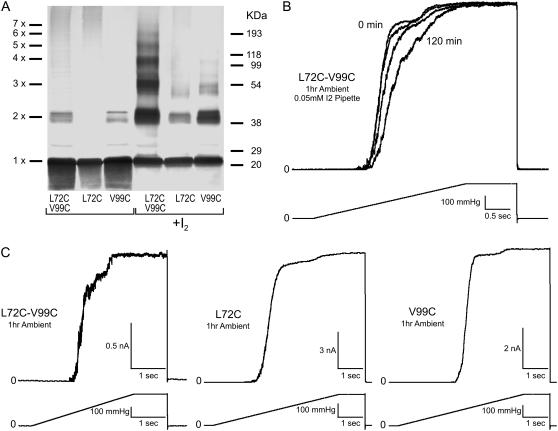
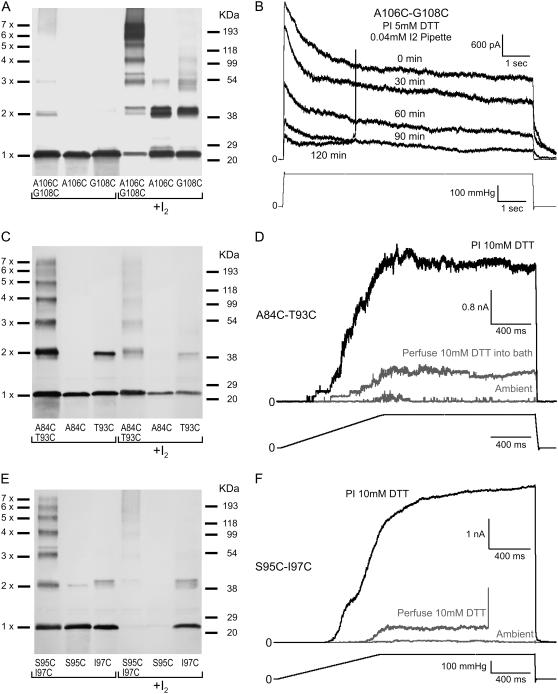
Channels from the MscS family are adaptive tension-activated osmolyte release valves that regulate turgor in prokaryotes and volume in plant chloroplasts. The crystal structure of Escherichia coli MscS has provided a starting point for detailed descriptions of its mechanism. However, solved in the absence of the lipid bilayer, this structure may deviate from a native conformation. In this study, we utilized molecular dynamics simulations and a new iterative extrapolated-motion protocol to pack the splayed peripheral TM1 and TM2 transmembrane helices along the central TM3 shaft. This modification restored the tension transmission route between the membrane and the channel gate. We also modeled the structure of the 26-amino acid N-terminal segments that were unresolved in the crystals. The resulting compact conformation, which we believe approximates the closed resting state of MscS, matches the hydrophobic thickness of the lipid bilayer with arginines 46, 54, and 74 facing the polar lipid headgroups. The pore-lining helices in this resting state feature alternative kinks near the conserved G121 instead of the G113 kinks observed in the crystal structure and the transmembrane barrel remains stable in extended molecular dynamics simulations. Further analysis of the dynamics of the pore constriction revealed several moderately asymmetric and largely dehydrated states. Biochemical and patch-clamp experiments with engineered double-cysteine mutants demonstrated cross-linking between predicted adjacent residue pairs, which formed either spontaneously or under moderate oxidation. The L72C-V99C bridge linking more peripheral TM2 to TM3 caused a shift of channel activation to higher pressures. TM3 to TM3 cross-links through the A84C-T93C, S95C-I97C, and A106C-G108C cysteine pairs were shown to lock MscS in a nonconductive state. Normal channel activity in these mutants could be recovered upon disulfide reduction with dithiothreitol. These results confirmed our modeling predictions of a closed MscS channel featuring a TM3 barrel that largely resembles the crystal conformation though with more tightly packed peripheral helices. From this closed-resting conformation, the TM3 helices must expand to allow for channel opening.
Figures
References
-
- Haswell, E. S., and E. M. Meyerowitz. 2006. MscS-like proteins control plastid size and shape in Arabidopsis thaliana. Curr. Biol. 16:1–11. - PubMed
-
- Okada, K., P. C. Moe, and P. Blount. 2002. Functional design of bacterial mechanosensitive channels. Comparisons and contrasts illuminated by random mutagenesis. J. Biol. Chem. 277:27682–27688. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
