

Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion
- PMID: 17992256
- PMCID: PMC2066199
- DOI: 10.1172/JCI32994
Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion
Abstract
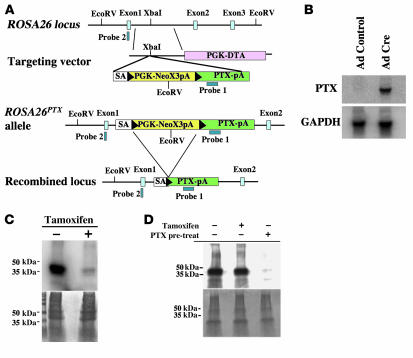
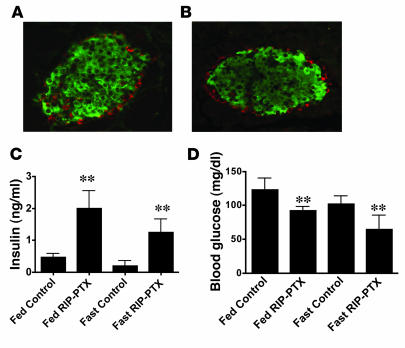
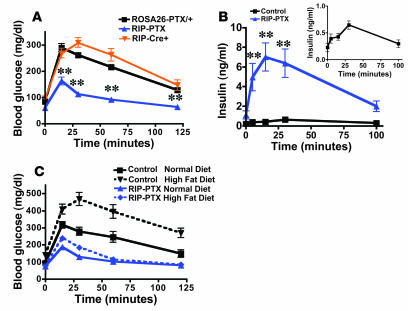
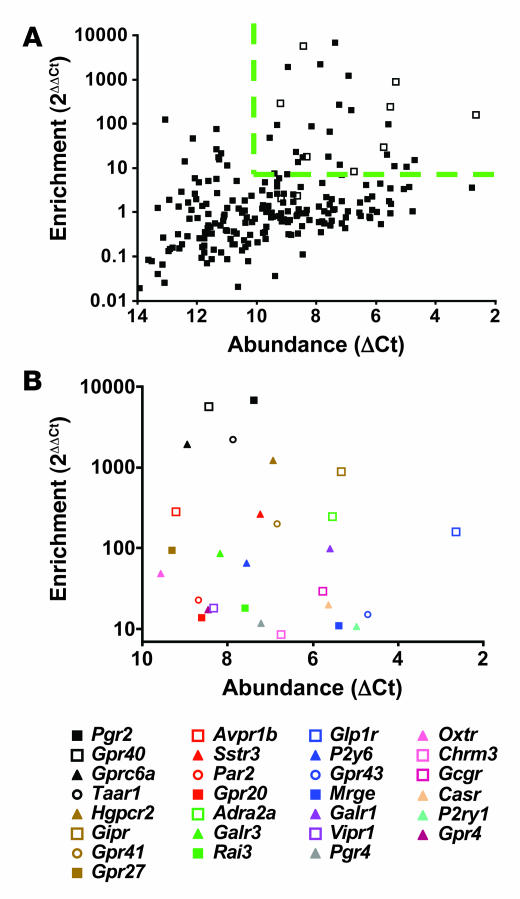
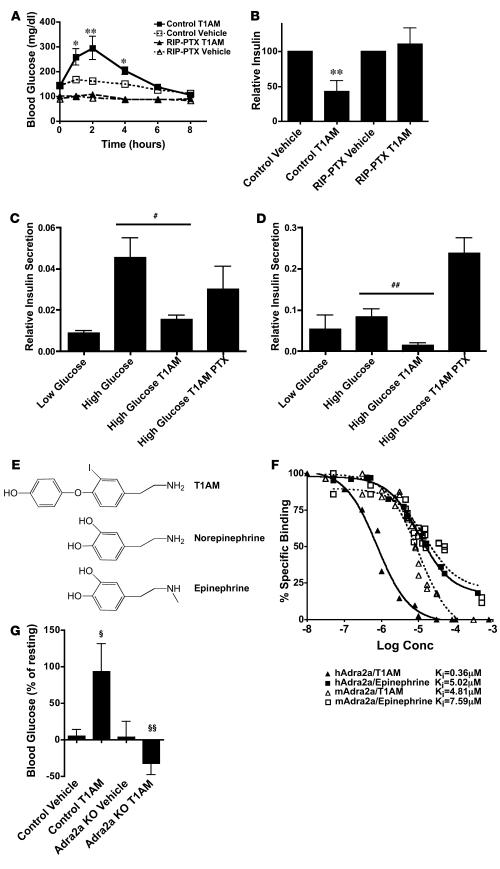
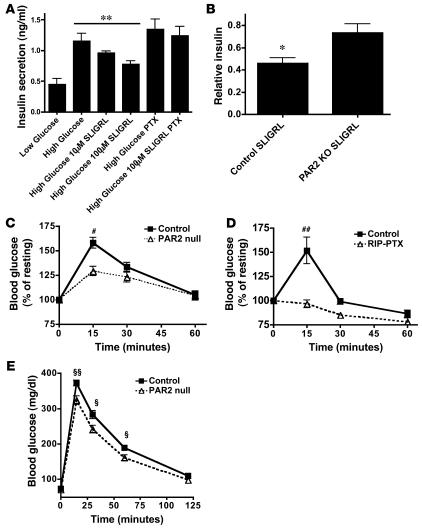
The in vivo roles of the hundreds of mammalian G protein-coupled receptors (GPCRs) are incompletely understood. To explore these roles, we generated mice expressing the S1 subunit of pertussis toxin, a known inhibitor of G(i/o) signaling, under the control of the ROSA26 locus in a Cre recombinase-dependent manner (ROSA26(PTX)). Crossing ROSA26(PTX) mice to mice expressing Cre in pancreatic beta cells produced offspring with constitutive hyperinsulinemia, increased insulin secretion in response to glucose, and resistance to diet-induced hyperglycemia. This phenotype underscored the known importance of G(i/o) and hence of GPCRs for regulating insulin secretion. Accordingly, we quantified mRNA for each of the approximately 373 nonodorant GPCRs in mouse to identify receptors highly expressed in islets and examined the role of several. We report that 3-iodothyronamine, a thyroid hormone metabolite, could negatively and positively regulate insulin secretion via the G(i)-coupled alpha(2A)-adrenergic receptor and the G(s)-coupled receptor Taar1, respectively, and protease-activated receptor-2 could negatively regulate insulin secretion and may contribute to physiological regulation of glucose metabolism. The ROSA26(PTX) system used in this study represents a new genetic tool to achieve tissue-specific signaling pathway modulation in vivo that can be applied to investigate the role of G(i/o)-coupled GPCRs in multiple cell types and processes.
Figures
References
-
- Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. - PubMed
-
- Civelli O. GPCR deorphanizations: the novel, the known and the unexpected transmitters. Trends Pharmacol. Sci. 2005;26:15–19. - PubMed
-
- Offermanns S., Mancino V., Revel J.P., Simon M.I. Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science. 1997;275:533–536. - PubMed
-
- Offermanns S., Toombs C.F., Hu Y.H., Simon M.I. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997;389:183–186. - PubMed
-
- Rudolph U., et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat. Genet. 1995;10:143–150. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
