

TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice
- PMID: 17992258
- PMCID: PMC2066188
- DOI: 10.1172/JCI32567
TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice
Abstract
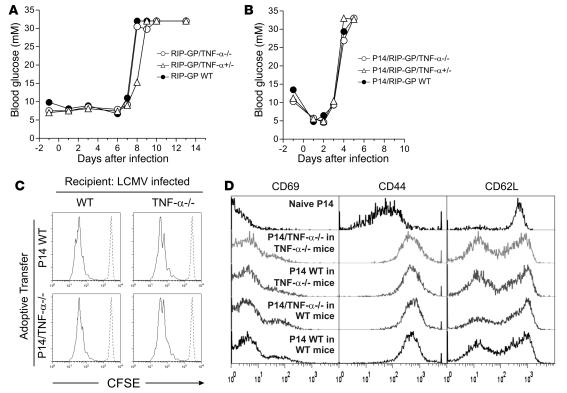
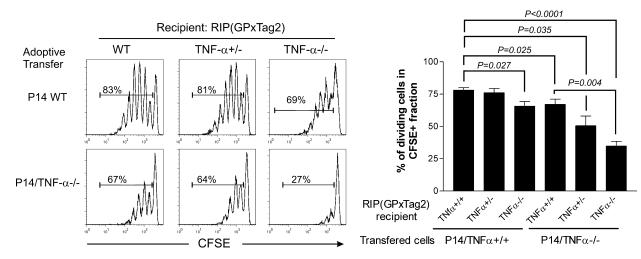
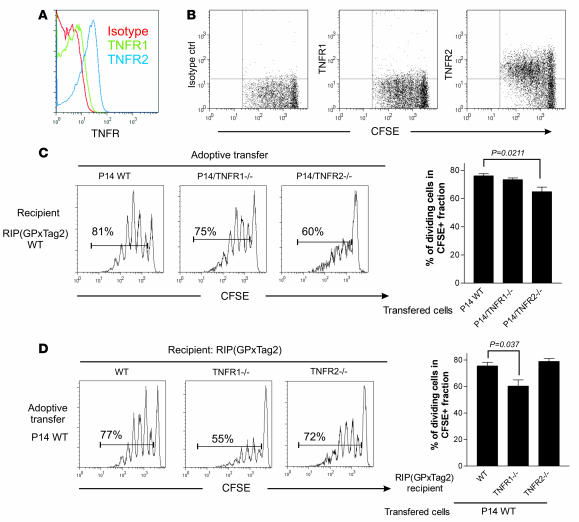
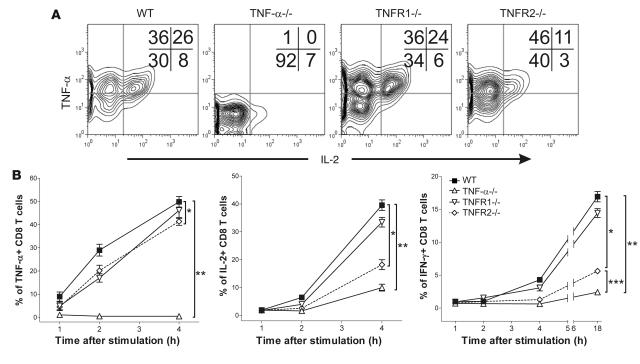
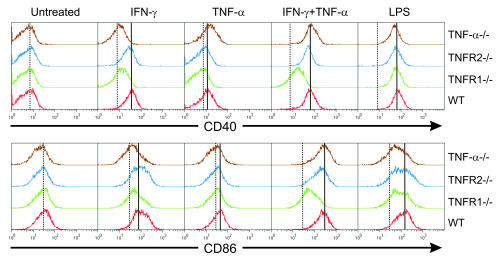
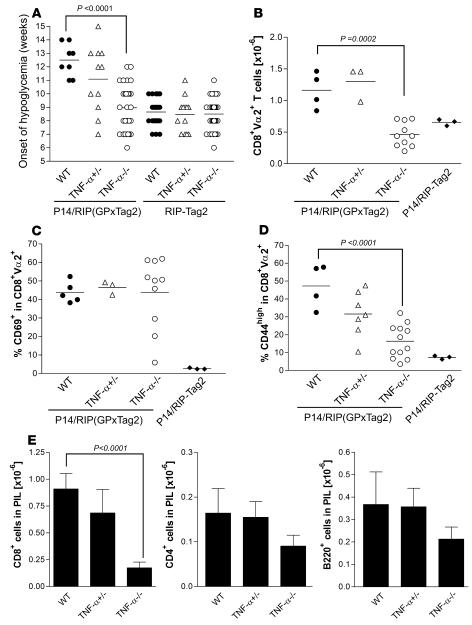
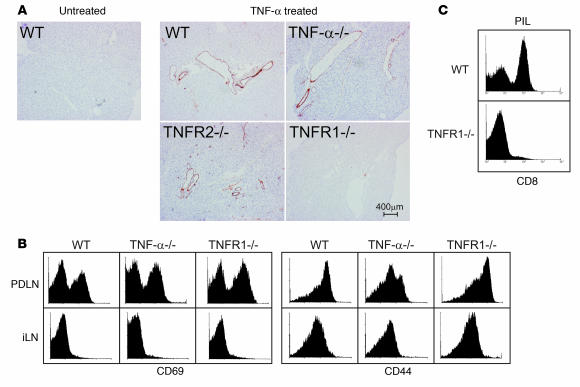
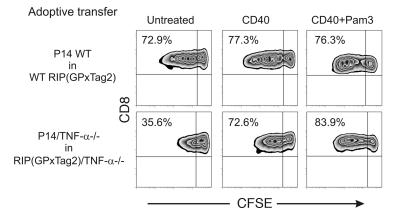
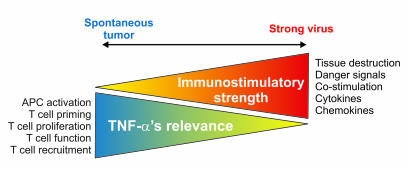
TNF-alpha antagonists are widely used in the treatment of inflammatory and autoimmune diseases, but their use is associated with reactivation of latent infections. This highlights the importance of TNF-alpha in immunity to certain pathogens and raises concerns that critical aspects of immune function are impaired in its absence. Unfortunately, the role of TNF-alpha in the regulation of T cell responses is clouded by a myriad of contradictory reports. Here, we show a role for TNF-alpha and its receptors, TNFR1 and TNFR2, specifically in antitumor immunity. TNF-alpha-deficient mice exhibited normal antiviral responses associated with strong inflammation. However, TNF-alpha/TNFR1-mediated signals on APCs and TNF-alpha/TNFR2 signals on T cells were critically required for effective priming, proliferation, and recruitment of tumor-specific T cells. Furthermore, in the absence of TNF-alpha signaling, tumor immune surveillance was severely abrogated. Finally, treatment with a CD40 agonist alone or in combination with TLR2 stimuli was able to rescue proliferation of TNF-alpha-deficient T cells. Therefore, TNF-alpha signaling may be required only for immune responses in conditions of limited immunostimulatory capacity, such as tumor surveillance. Importantly, these results suggest that prolonged continuous TNF-alpha blockade in patients may have long-term complications, including potential tumor development or progression.
Figures
References
-
- MacEwan D.J. TNF receptor subtype signalling: differences and cellular consequences. Cell Signal. 2002;14:477–492. - PubMed
-
- Wajant H., Pfizenmaier K., Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. - PubMed
-
- Kollias G. TNF pathophysiology in murine models of chronic inflammation and autoimmunity. Semin. Arthritis Rheum. 2005;34:3–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
