

Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data
- PMID: 17993489
- PMCID: PMC2242766
- DOI: 10.1529/biophysj.107.122218
Normal-mode flexible fitting of high-resolution structure of biological molecules toward one-dimensional low-resolution data
Abstract
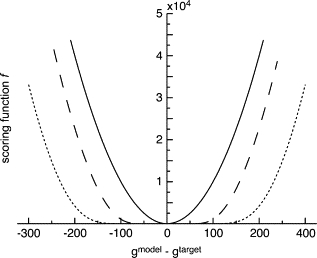
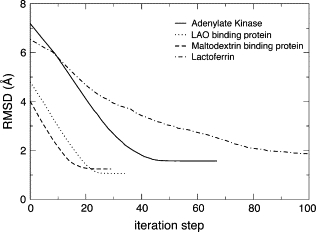
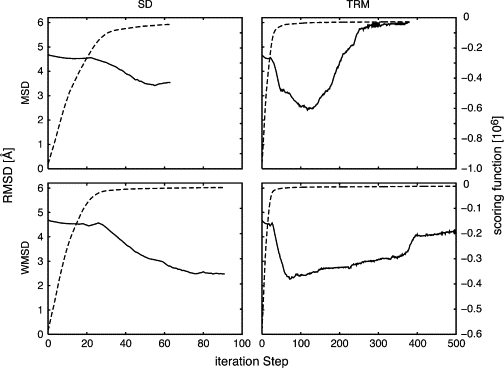
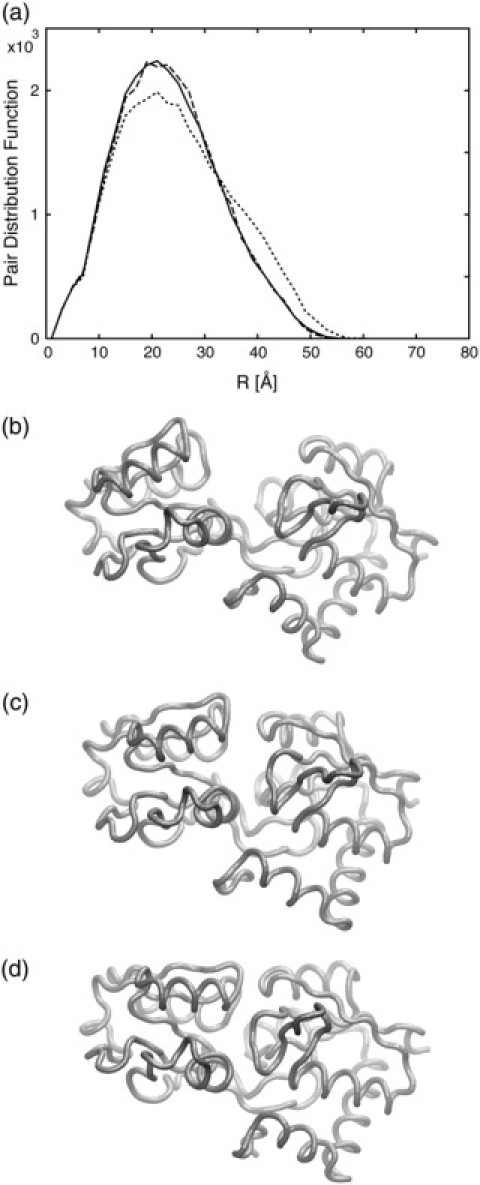
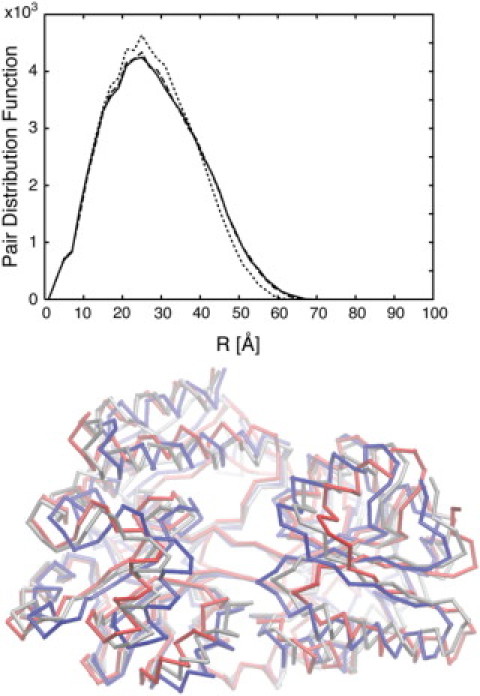
We present a method for reconstructing a 3D structure from a pair distribution function by flexibly fitting known x-ray structures toward a conformation that agrees with the low-resolution data. This method uses a linear combination of low-frequency normal modes from elastic-network description of the molecule in an iterative manner to deform the structure optimally to conform to the target pair distribution function. A simple function, pair distance distribution function between atoms, is chosen as a test model to establish computational algorithms-optimization algorithm and scoring function-that can utilize low-resolution 1D data. To select a correct structural model based on less information, we developed a scoring function that takes into account a characteristic of pair distribution functions. In addition, we employ a new optimization algorithm, the trusted region method, that relies on both first and second derivatives of the scoring function. Illustrative results of our studies on simulated 1D data from five different proteins, for which large conformational changes are known to occur, are presented.
Figures

References
-
- Saibil H.R. Conformational changes studied by cryo-electron microscopy. Nat. Struct. Biol. 2000;7:711–714. - PubMed
-
- Heller W.T., Vigil D., Brown S., Blumenthal D.K., Taylor S.S., Trewhella J. C subunits binding to the protein kinase A RIα dimer induce a large conformational change. J. Biol. Chem. 2004;279:19084–19090. - PubMed
-
- Vestergaard B., Sanyal S., Roessle M., Mora L., Buckingham R.H., Kastrup J.S., Gajhede M., Svergun D.I., Ehrenberg M. The SAXS solution structure of RF1 differs from its crystal structure and is similar to its ribosome bound cryo-EM structure. Mol. Cell. 2005;20:929–938. - PubMed
-
- Ermolenko D.N., Spiegel P.C., Majumdar Z.K., Hickerson R.P., Clegg R.M., Noller H.F. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat. Struct. Mol. Biol. 2007;14:493–497. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
