

Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice
- PMID: 17993608
- PMCID: PMC2254536
- DOI: 10.1182/blood-2007-08-109710
Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice
Abstract
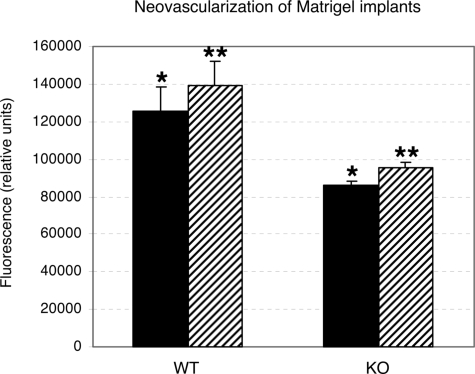
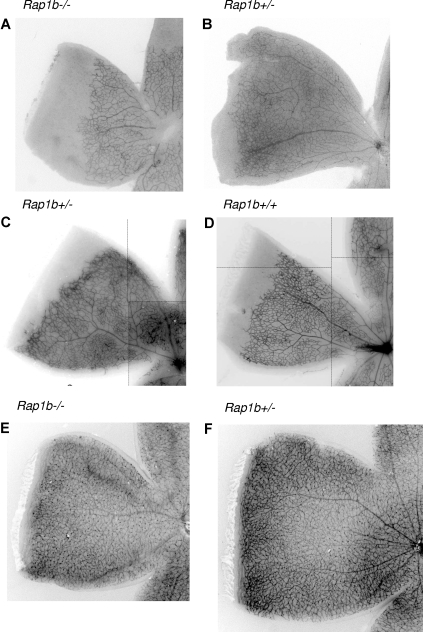
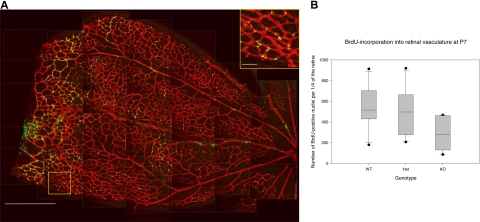
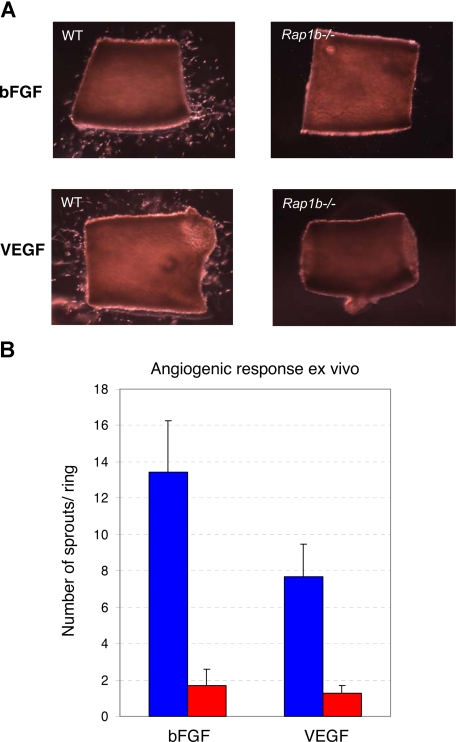
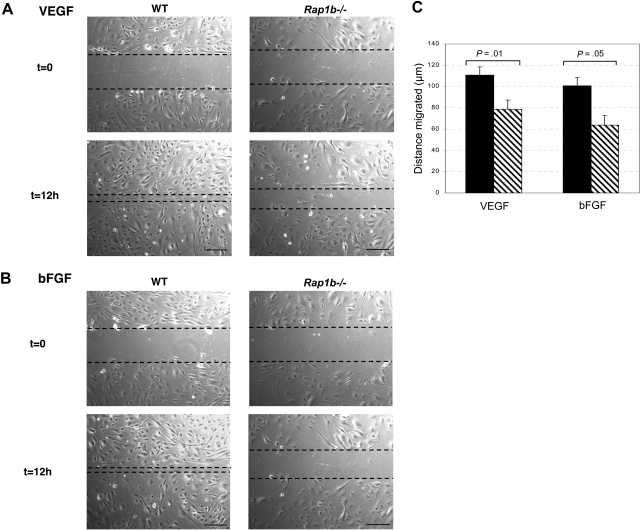
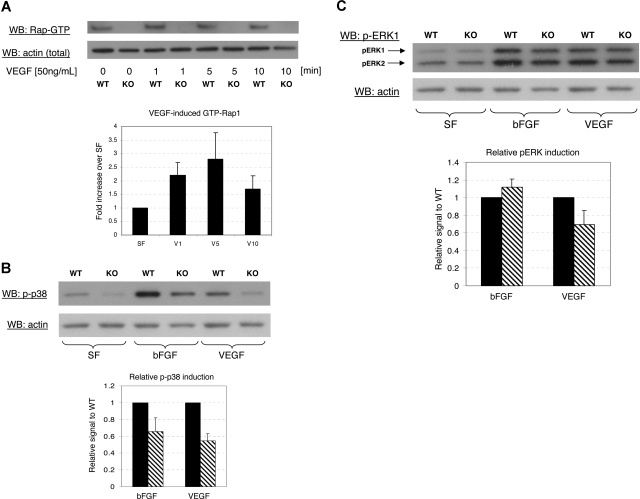
Angiogenesis is the main mechanism of vascular remodeling during late development and, after birth, in wound healing. Perturbations of angiogenesis occur in cancer, diabetes, ischemia, and inflammation. While much progress has been made in identifying factors that control angiogenesis, the understanding of the precise molecular mechanisms involved is incomplete. Here we identify a small GTPase, Rap1b, as a positive regulator of angiogenesis. Rap1b-deficient mice had a decreased level of Matrigel plug and neonatal retinal neovascularization, and aortas isolated from Rap1b-deficient animals had a reduced microvessel sprouting response to 2 major physiological regulators of angiogenesis: vascular endothelial growth factor (VEGF) and basic fibroblasts growth factor (bFGF), indicating an intrinsic defect in endothelial cells. Proliferation of retinal endothelial cells in situ and in vitro migration of lung endothelial cells isolated from Rap1b-deficient mice were inhibited. At the molecular level, activation of 2 MAP kinases, p38 MAPK and p42/44 ERK, important regulators of endothelial migration and proliferation, was decreased in Rap1b-deficient endothelial cells in response to VEGF stimulation. These studies provide evidence that Rap1b is required for normal angiogenesis and reveal a novel role of Rap1 in regulation of proangiogenic signaling in endothelial cells.
Figures

Similar articles
-
Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions.Mol Cell Biol. 2008 Sep;28(18):5803-10. doi: 10.1128/MCB.00393-08. Epub 2008 Jul 14. Mol Cell Biol. 2008. PMID: 18625726 Free PMC article.
-
Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin αvβ₃.Blood. 2011 Aug 18;118(7):2015-26. doi: 10.1182/blood-2011-04-349282. Epub 2011 Jun 2. Blood. 2011. PMID: 21636859 Free PMC article.
-
Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis.Blood. 2009 Jan 8;113(2):488-97. doi: 10.1182/blood-2008-02-138438. Epub 2008 Sep 19. Blood. 2009. PMID: 18805968
-
Towards Targeting Endothelial Rap1B to Overcome Vascular Immunosuppression in Cancer.Int J Mol Sci. 2024 Sep 12;25(18):9853. doi: 10.3390/ijms25189853. Int J Mol Sci. 2024. PMID: 39337337 Free PMC article. Review.
-
Divergent Functions of Rap1A and Rap1B in Endothelial Biology and Disease.Int J Mol Sci. 2025 Jun 4;26(11):5372. doi: 10.3390/ijms26115372. Int J Mol Sci. 2025. PMID: 40508181 Free PMC article. Review.
Cited by
-
Endothelial Rap1B mediates T-cell exclusion to promote tumor growth: a novel mechanism underlying vascular immunosuppression.Angiogenesis. 2023 May;26(2):265-278. doi: 10.1007/s10456-022-09862-5. Epub 2022 Nov 20. Angiogenesis. 2023. PMID: 36403190
-
Inhibitory effects of Rap1GAP overexpression on proliferation and migration of endothelial cells via ERK and Akt pathways.J Huazhong Univ Sci Technolog Med Sci. 2011 Dec;31(6):721-727. doi: 10.1007/s11596-011-0667-x. Epub 2011 Dec 16. J Huazhong Univ Sci Technolog Med Sci. 2011. PMID: 22173489
-
TRPM8 and prostate: a cold case?Pflugers Arch. 2018 Oct;470(10):1419-1429. doi: 10.1007/s00424-018-2169-1. Epub 2018 Jun 20. Pflugers Arch. 2018. PMID: 29926226 Review.
-
Hematopoietic protein tyrosine phosphatase mediates beta2-adrenergic receptor-induced regulation of p38 mitogen-activated protein kinase in B lymphocytes.Mol Cell Biol. 2009 Feb;29(3):675-86. doi: 10.1128/MCB.01466-08. Epub 2008 Dec 1. Mol Cell Biol. 2009. PMID: 19047375 Free PMC article.
-
Glioma-associated endothelial cells are chemoresistant to temozolomide.J Neurooncol. 2009 Oct;95(1):13-22. doi: 10.1007/s11060-009-9891-7. Epub 2009 Apr 18. J Neurooncol. 2009. PMID: 19381445
References
-
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. - PubMed
-
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. - PubMed
-
- Shalaby F, Rossant J, Yamaguchi TP, et al. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. - PubMed
-
- Rousseau S, Houle F, Landry J, Huota J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
