

Mulan: multiple-sequence alignment to predict functional elements in genomic sequences
- PMID: 17993678
- PMCID: PMC3704129
Mulan: multiple-sequence alignment to predict functional elements in genomic sequences
Abstract
Multiple sequence alignment analysis is a powerful approach for translating the evolutionary selective power into phylogenetic relationships to localize functional coding and noncoding genomic elements. The tool Mulan (http://mulan.dcode.org/) has been designed to effectively perform multiple comparisons of genomic sequences necessary to facilitate bioinformatic-driven biological discoveries. The Mulan network server is capable of comparing both closely and distantly related genomes to identify conserved elements over a broad range of evolutionary time. Several novel algorithms are brought together in this tool: the tba multisequence aligner program used to rapidly identify local sequence conservation and the multiTF program to detect evolutionarily conserved transcription factor binding sites in alignments. Mulan is integrated with the ERC Browser, the UCSC Genome Browser for quick uploads of available sequences and supports two-way communication with the GALA database to overlay GALA functional genome annotation with sequence conservation profiles. Local multiple alignments computed by Mulan ensure reliable representation of short- and large-scale genomic rearrangements in distant organisms. Recently, we have also introduced the ability to handle duplications to permit the reliable reconstruction of evolutionary events that underlie the genome sequence data. Here, we describe the main features of the Mulan tool that include the interactive modification of critical conservation parameters, visualization options, and dynamic access to sequence data from visual graphs for flexible and easy-to-perform analysis of differentially evolving genomic regions.
Figures




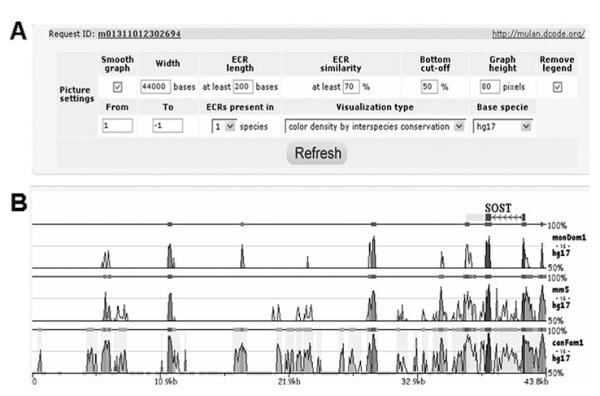
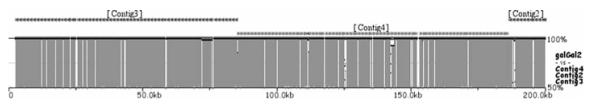
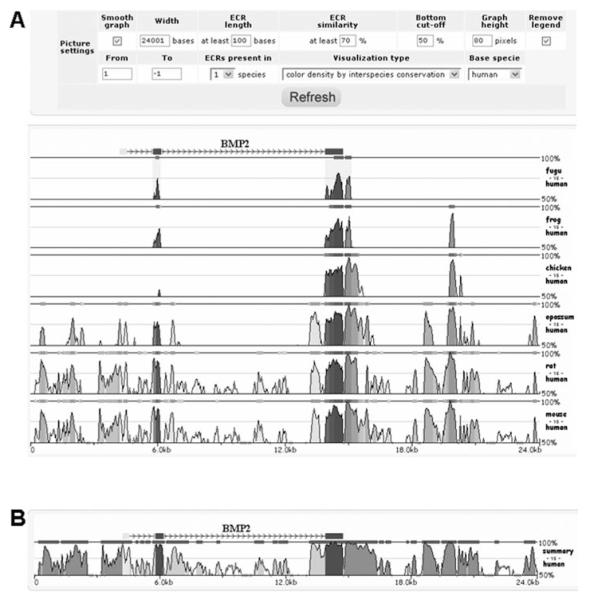

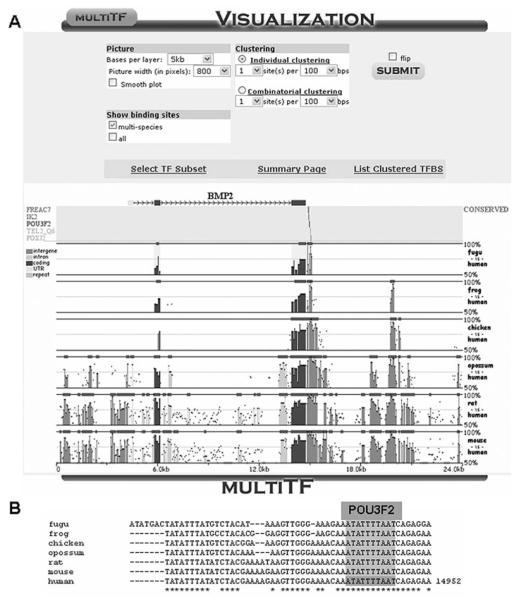
Similar articles
-
Mulan: multiple-sequence local alignment and visualization for studying function and evolution.Genome Res. 2005 Jan;15(1):184-94. doi: 10.1101/gr.3007205. Epub 2004 Dec 8. Genome Res. 2005. PMID: 15590941 Free PMC article.
-
Dcode.org anthology of comparative genomic tools.Nucleic Acids Res. 2005 Jul 1;33(Web Server issue):W56-64. doi: 10.1093/nar/gki355. Nucleic Acids Res. 2005. PMID: 15980535 Free PMC article.
-
webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser.BMC Bioinformatics. 2010 Nov 26;11:579. doi: 10.1186/1471-2105-11-579. BMC Bioinformatics. 2010. PMID: 21110866 Free PMC article.
-
Computational prediction of cis-regulatory modules from multispecies alignments using Galaxy, Table Browser, and GALA.Methods Mol Biol. 2006;338:91-103. doi: 10.1385/1-59745-097-9:91. Methods Mol Biol. 2006. PMID: 16888352
-
Multiple sequence alignment: in pursuit of homologous DNA positions.Genome Res. 2007 Feb;17(2):127-35. doi: 10.1101/gr.5232407. Genome Res. 2007. PMID: 17272647 Review.
Cited by
-
Spermatogenesis associated 4 promotes Sertoli cell proliferation modulated negatively by regulatory factor X1.PLoS One. 2013 Oct 11;8(10):e75933. doi: 10.1371/journal.pone.0075933. eCollection 2013. PLoS One. 2013. PMID: 24146794 Free PMC article.
-
Mechanism of upregulation of the inhibitory regulator, src-like adaptor protein (SLAP), by glucocorticoids in mast cells.Mol Immunol. 2009 Jan;46(3):492-7. doi: 10.1016/j.molimm.2008.10.011. Epub 2008 Nov 25. Mol Immunol. 2009. PMID: 19036452 Free PMC article.
-
Comparative analysis of the ATRX promoter and 5' regulatory region reveals conserved regulatory elements which are linked to roles in neurodevelopment, alpha-globin regulation and testicular function.BMC Res Notes. 2011 Jun 15;4:200. doi: 10.1186/1756-0500-4-200. BMC Res Notes. 2011. PMID: 21676266 Free PMC article.
-
Early B-cell factor-1 (EBF1) is a key regulator of metabolic and inflammatory signaling pathways in mature adipocytes.J Biol Chem. 2013 Dec 13;288(50):35925-39. doi: 10.1074/jbc.M113.491936. Epub 2013 Oct 30. J Biol Chem. 2013. PMID: 24174531 Free PMC article.
-
Use of ChIP-Seq data for the design of a multiple promoter-alignment method.Nucleic Acids Res. 2012 Apr;40(7):e52. doi: 10.1093/nar/gkr1292. Epub 2012 Jan 9. Nucleic Acids Res. 2012. PMID: 22230796 Free PMC article.
References
-
- Pennacchio LA, Olivier M, Hubacek JA, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. - PubMed
-
- Gilligan P, Brenner S, Venkatesh B. Fugu and human sequence comparison identifies novel human genes and conserved non-coding sequences. Gene. 2002;294:35–44. - PubMed
-
- Elnitski L, Li J, Noguchi CT, Miller W, Hardison R. A negative cis-element regulates the level of enhancement by hypersensitive site 2 of the beta-globin locus control region. J. Biol. Chem. 2001;276:6289–6298. - PubMed
-
- Loots GG, Locksley RM, Blankespoor CM, et al. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. - PubMed
-
- Mayor C, Brudno M, Schwartz JR, et al. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources