

Antiinflammatory and neuroprotective actions of COX2 inhibitors in the injured brain
- PMID: 17996418
- PMCID: PMC2855502
- DOI: 10.1016/j.bbi.2007.09.011
Antiinflammatory and neuroprotective actions of COX2 inhibitors in the injured brain
Abstract
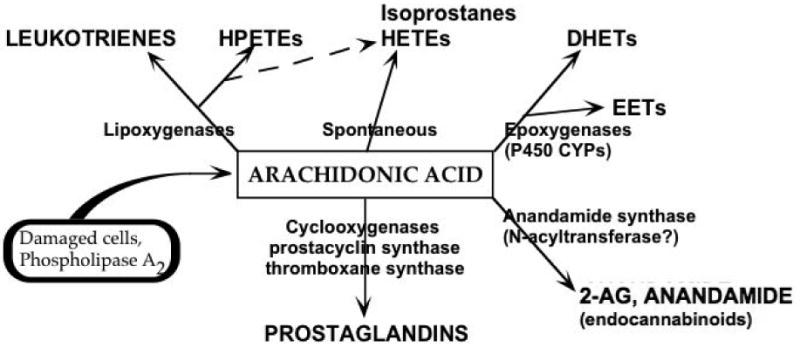
Overexpression of COX2 appears to be both a marker and an effector of neural damage after a variety of acquired brain injuries, and in natural or pathological aging of the brain. COX2 inhibitors may be neuroprotective in the brain by reducing prostanoid and free radical synthesis, or by directing arachidonic acid down alternate metabolic pathways. The arachidonic acid shunting hypothesis proposes that COX2 inhibitors' neuroprotective effects may be mediated by increased formation of potentially beneficial eicosanoids. Under conditions where COX2 activity is inhibited, arachidonic acid accumulates or is converted to eicosanoids via lipoxygenases and cytochrome P450 (CYP) epoxygenases. Several P450 eicosanoids have been demonstrated to have beneficial effects in the brain and/or periphery. We suspect that arachidonic acid shunting may be as important to functional recovery after brain injuries as altered prostanoid formation per se. Thus, COX2 inhibition and arachidonic acid shunting have therapeutic implications beyond the suppression of prostaglandin synthesis and free radical formation.
Figures

Similar articles
-
Regulation of inflammation in cancer by eicosanoids.Prostaglandins Other Lipid Mediat. 2011 Nov;96(1-4):27-36. doi: 10.1016/j.prostaglandins.2011.08.004. Epub 2011 Aug 16. Prostaglandins Other Lipid Mediat. 2011. PMID: 21864702 Free PMC article. Review.
-
Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury.Neurosurgery. 2005 Mar;56(3):590-604. doi: 10.1227/01.neu.0000154060.14900.8f. Neurosurgery. 2005. PMID: 15730585 Free PMC article.
-
Acute arsenic treatment alters arachidonic acid and its associated metabolite levels in the brain of C57Bl/6 mice.Can J Physiol Pharmacol. 2014 Aug;92(8):693-702. doi: 10.1139/cjpp-2014-0136. Can J Physiol Pharmacol. 2014. PMID: 25065748
-
Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation.Science. 2011 Nov 11;334(6057):809-13. doi: 10.1126/science.1209200. Epub 2011 Oct 20. Science. 2011. PMID: 22021672 Free PMC article.
-
Cytochrome P450-derived eicosanoids: the neglected pathway in cancer.Cancer Metastasis Rev. 2010 Dec;29(4):723-35. doi: 10.1007/s10555-010-9264-x. Cancer Metastasis Rev. 2010. PMID: 20941528 Free PMC article. Review.
Cited by
-
Emerging therapeutic targets for cerebral edema.Expert Opin Ther Targets. 2021 Nov;25(11):917-938. doi: 10.1080/14728222.2021.2010045. Epub 2022 Jan 2. Expert Opin Ther Targets. 2021. PMID: 34844502 Free PMC article. Review.
-
Cyclooxygenase 2 Inhibitors for Headache After Elective Cranial Neurosurgery: Results from a Systematic Review of Efficacy of Cyclooxygenase 2 Inhibitors for Headache After Acute Brain Injury.Neurocrit Care. 2025 Apr;42(2):680-689. doi: 10.1007/s12028-024-02114-y. Epub 2024 Sep 12. Neurocrit Care. 2025. PMID: 39266865
-
Oxidative and nitrative stress in neurodegeneration.Neurobiol Dis. 2015 Dec;84:4-21. doi: 10.1016/j.nbd.2015.04.020. Epub 2015 May 27. Neurobiol Dis. 2015. PMID: 26024962 Free PMC article. Review.
-
Intra-amniotic LPS causes acute neuroinflammation in preterm rhesus macaques.J Neuroinflammation. 2016 Sep 6;13(1):238. doi: 10.1186/s12974-016-0706-4. J Neuroinflammation. 2016. PMID: 27596440 Free PMC article.
-
Cyclooxygenase (COX)-1 activity precedes the COX-2 induction in Aβ-induced neuroinflammation.J Mol Neurosci. 2011 Sep;45(1):10-21. doi: 10.1007/s12031-010-9401-6. Epub 2010 Jun 12. J Mol Neurosci. 2011. PMID: 20549385
References
-
- Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. - PubMed
-
- Acarin L, Gonzalez B, Castellano B. Triflusal posttreatment inhibits glial nuclear factor-kappaB, downregulates the glial response, and is neuroprotective in an excitotoxic injury model in postnatal brain. Stroke. 2001;32:2394–2402. - PubMed
-
- Acarin L, G B, C B. Decrease of proinflammatory molecules correlates with neuroprotective effect of the fluorinated salicylate triflusal after postnatal excitotoxic damage. Stroke. 2002;33:2499–2505. - PubMed
-
- Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. Journal of Neurochemistry. 1996;66:6–13. - PubMed
-
- Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, Hurn PD. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke. 2002;33:1677–1684. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
