

Genetic progression and the waiting time to cancer
- PMID: 17997597
- PMCID: PMC2065895
- DOI: 10.1371/journal.pcbi.0030225
Genetic progression and the waiting time to cancer
Abstract
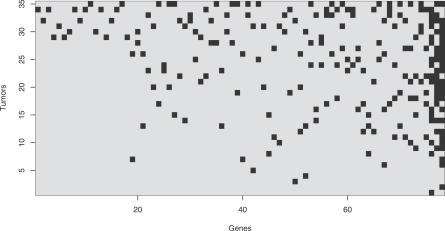
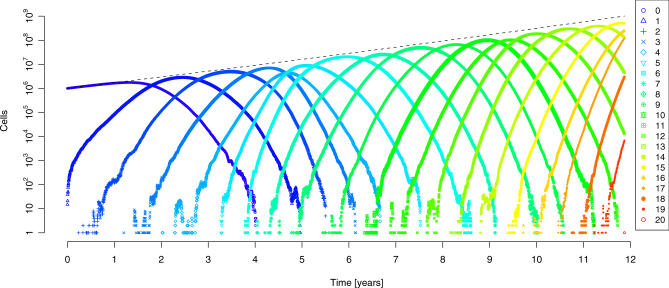
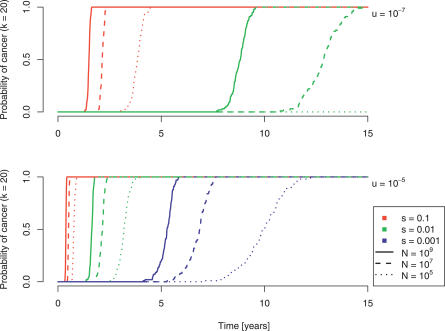
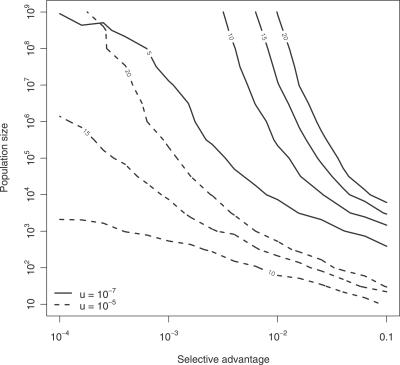
Cancer results from genetic alterations that disturb the normal cooperative behavior of cells. Recent high-throughput genomic studies of cancer cells have shown that the mutational landscape of cancer is complex and that individual cancers may evolve through mutations in as many as 20 different cancer-associated genes. We use data published by Sjöblom et al. (2006) to develop a new mathematical model for the somatic evolution of colorectal cancers. We employ the Wright-Fisher process for exploring the basic parameters of this evolutionary process and derive an analytical approximation for the expected waiting time to the cancer phenotype. Our results highlight the relative importance of selection over both the size of the cell population at risk and the mutation rate. The model predicts that the observed genetic diversity of cancer genomes can arise under a normal mutation rate if the average selective advantage per mutation is on the order of 1%. Increased mutation rates due to genetic instability would allow even smaller selective advantages during tumorigenesis. The complexity of cancer progression can be understood as the result of multiple sequential mutations, each of which has a relatively small but positive effect on net cell growth.
Conflict of interest statement
Figures

References
-
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Knudson AG. Cancer genetics. Am J Med Genet. 2002;111:96–102. - PubMed
-
- Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
