

Expression of a Cu,Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of neuroblastoma cells to infectious injury
- PMID: 17997855
- PMCID: PMC2211486
- DOI: 10.1186/1471-2334-7-131
Expression of a Cu,Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of neuroblastoma cells to infectious injury
Abstract
Background: Infections can aggravate the course of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). Mutations in the anti-oxidant enzyme Cu,Zn superoxide dismutase (EC 1.15.1.1, SOD1) are associated with familial ALS. Streptococcus pneumoniae, the most frequent respiratory pathogen, causes damage by the action of the cholesterol-binding virulence factor pneumolysin and by stimulation of the innate immune system, particularly via Toll-like-receptor 2.
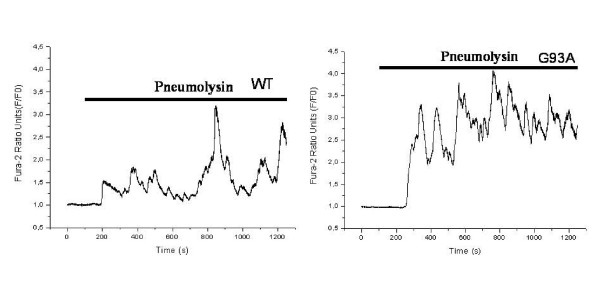
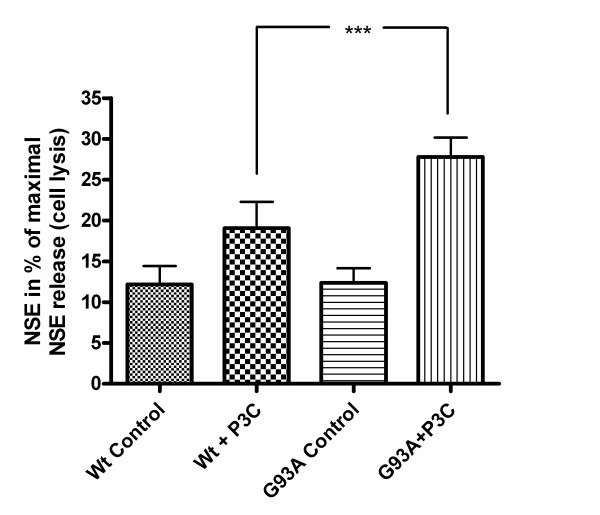
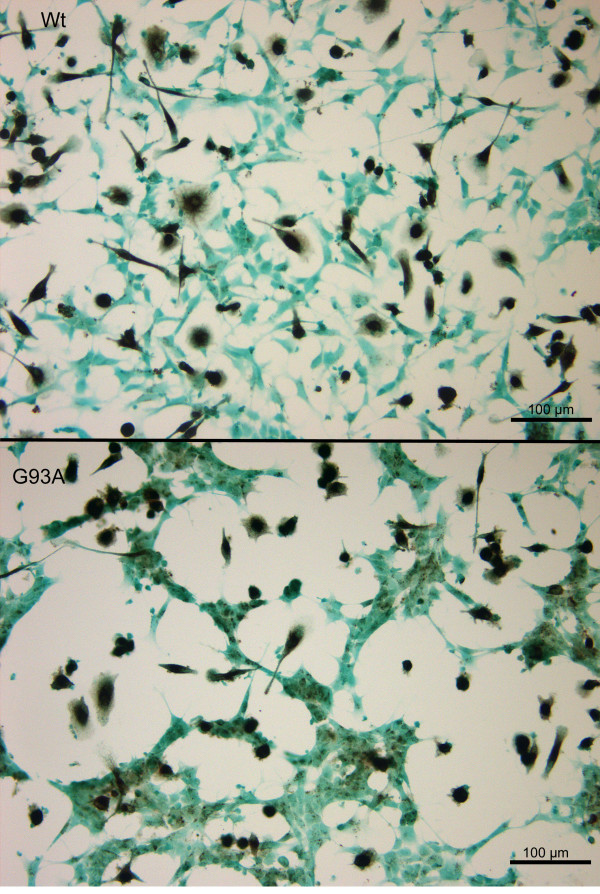
Methods: SH-SY5Y neuroblastoma cells transfected with the G93A mutant of SOD1 typical for familial ALS (G93A-SOD1) and SH-SY5Y neuroblastoma cells transfected with wildtype SOD1 were both exposed to pneumolysin and in co-cultures with cultured human macrophages treated with the Toll like receptor 2 agonist N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2RS)-propyl]-[R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine x 3 HCl (Pam3CSK4). Cell viability and apoptotic cell death were compared morphologically and by in-situ tailing. With the help of the WST-1 test, cell viability was quantified, and by measurement of neuron-specific enolase in the culture supernatant neuronal damage in co-cultures was investigated. Intracellular calcium levels were measured by fluorescence analysis using fura-2 AM.
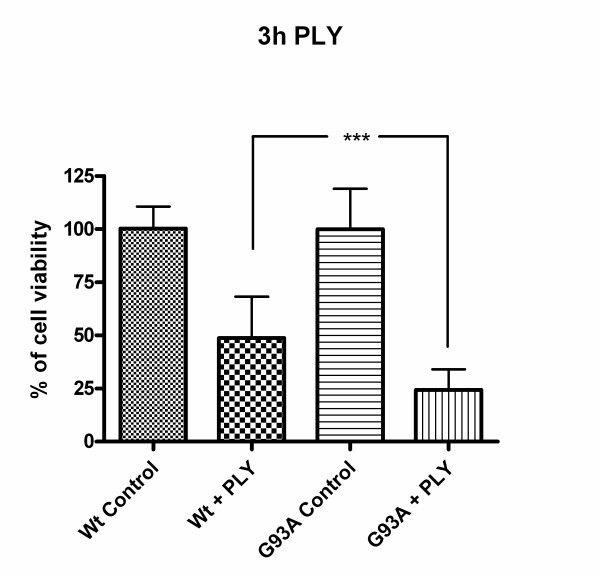
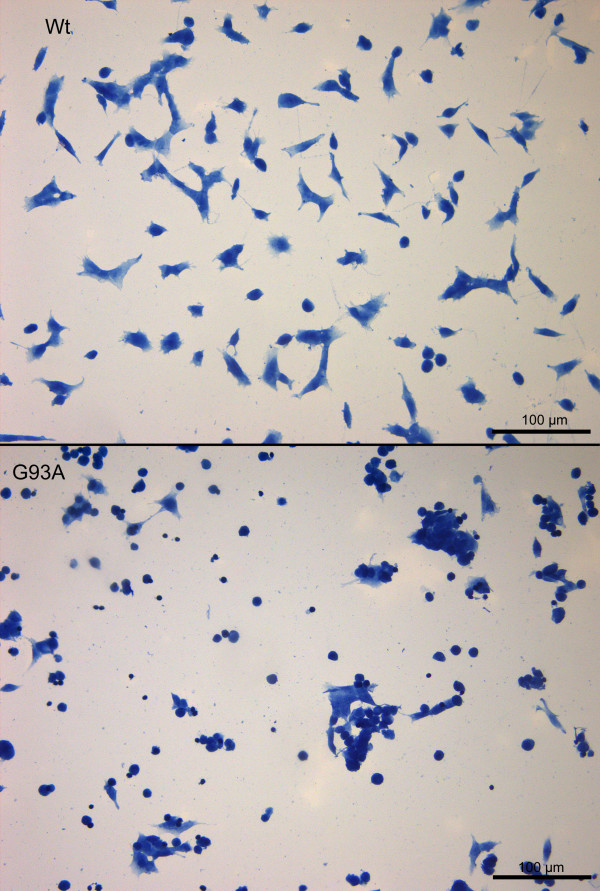
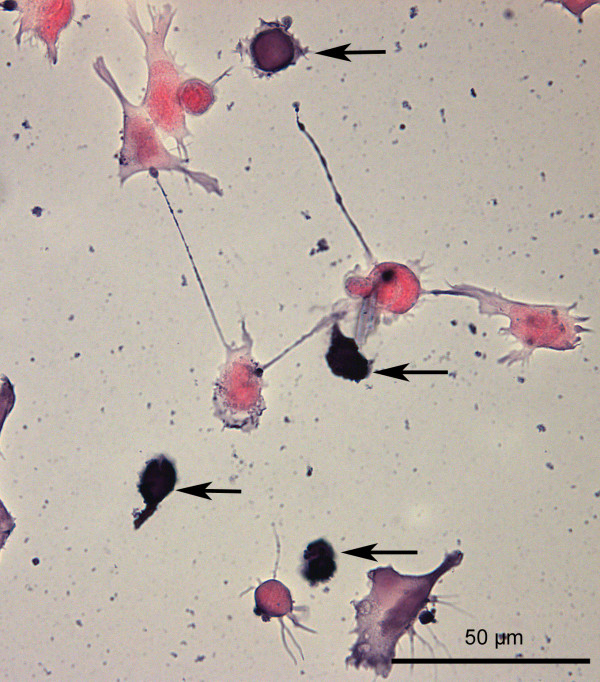
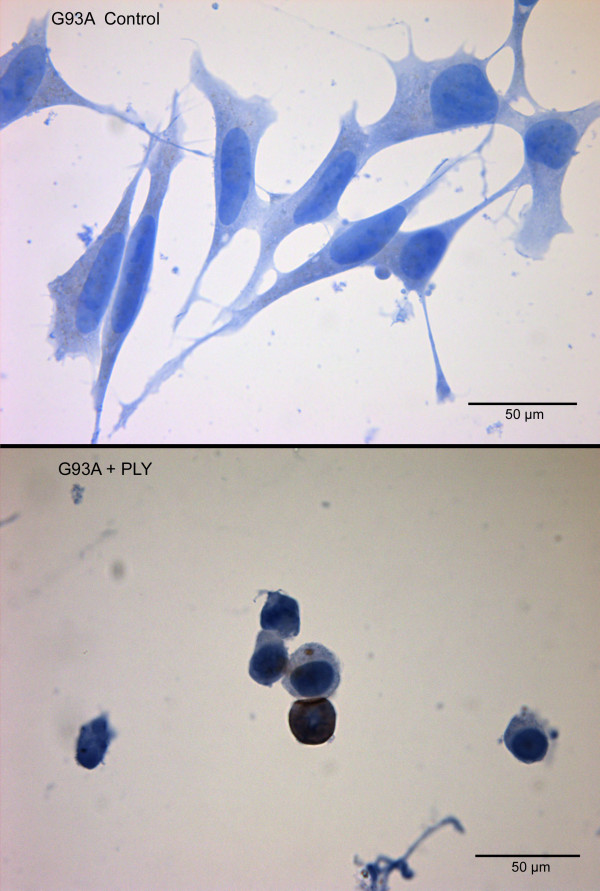
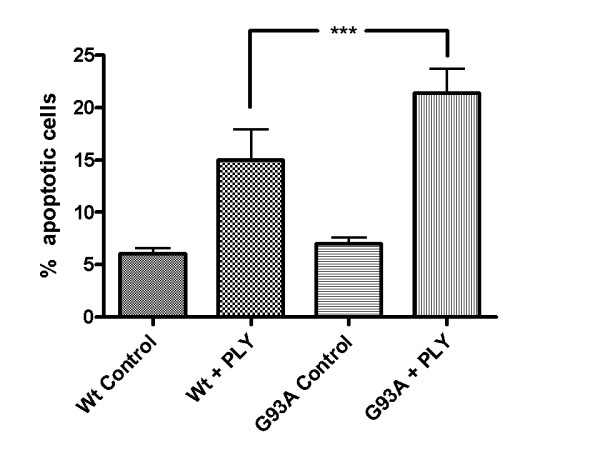
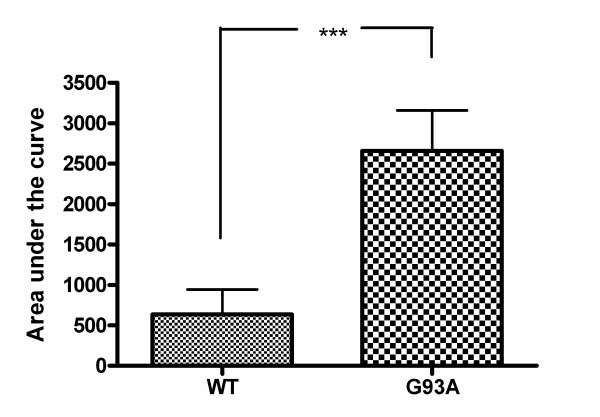
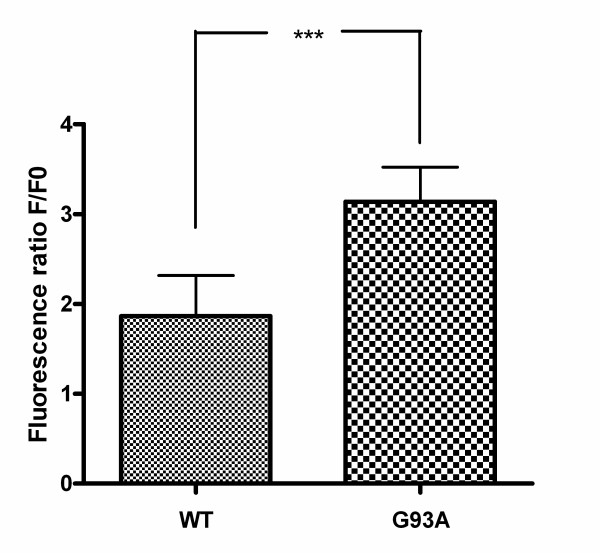
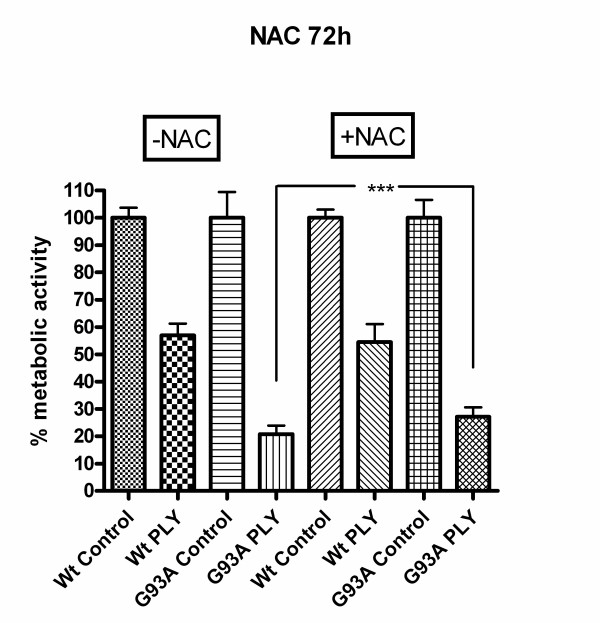
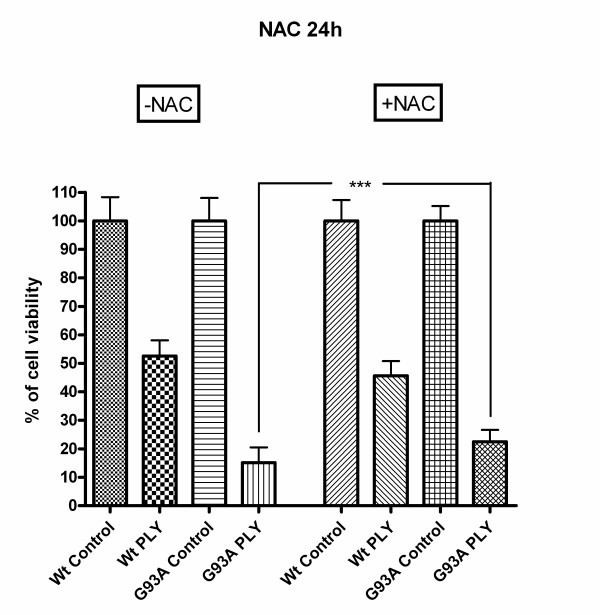
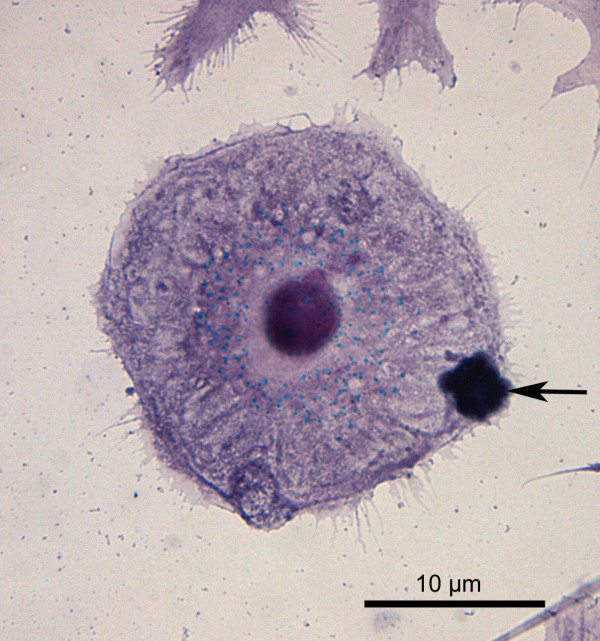
Results: SH-SY5Y neuroblastoma cells transfected with the G93A mutant of SOD1 typical for familial ALS (G93A-SOD1) were more vulnerable to the neurotoxic action of pneumolysin and to the attack of monocytes stimulated by Pam3CSK4 than SH-SY5Y cells transfected with wild-type human SOD1. The enhanced pneumolysin toxicity in G93A-SOD1 neuronal cells depended on the inability of these cells to cope with an increased calcium influx caused by pores formed by pneumolysin. This inability was caused by an impaired capacity of the mitochondria to remove cytoplasmic calcium. Treatment of G93A-SOD1 SH-SY5Y neuroblastoma cells with the antioxidant N-acetylcysteine reduced the toxicity of pneumolysin.
Conclusion: The particular vulnerability of G93A-SOD1 neuronal cells to hemolysins and inflammation may be partly responsible for the clinical deterioration of ALS patients during infections. These findings link infection and motor neuron disease and suggest early treatment of respiratory infections in ALS patients.
Figures
Similar articles
-
Impairment of glutamate transport and increased vulnerability to oxidative stress in neuroblastoma SH-SY5Y cells expressing a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis.Neurochem Int. 2005 Feb;46(3):227-34. doi: 10.1016/j.neuint.2004.10.002. Epub 2004 Dec 8. Neurochem Int. 2005. PMID: 15670639
-
Cu/Zn superoxide dismutase (SOD1) mutations associated with familial amyotrophic lateral sclerosis (ALS) affect cellular free radical release in the presence of oxidative stress.Amyotroph Lateral Scler Other Motor Neuron Disord. 2002 Jun;3(2):75-85. doi: 10.1080/146608202760196048. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002. PMID: 12215229
-
Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS.Biochim Biophys Acta. 2010 May;1802(5):462-71. doi: 10.1016/j.bbadis.2010.01.011. Epub 2010 Jan 25. Biochim Biophys Acta. 2010. PMID: 20097285
-
Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation.Neuropathology. 2001 Mar;21(1):82-92. doi: 10.1046/j.1440-1789.2001.00361.x. Neuropathology. 2001. PMID: 11304046 Review.
-
The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death.Neurochem Res. 2009 Oct;34(10):1847-56. doi: 10.1007/s11064-009-9974-z. Epub 2009 Apr 28. Neurochem Res. 2009. PMID: 19399611 Review.
Cited by
-
Roles of vitamin D in amyotrophic lateral sclerosis: possible genetic and cellular signaling mechanisms.Mol Brain. 2013 Apr 9;6:16. doi: 10.1186/1756-6606-6-16. Mol Brain. 2013. PMID: 23570271 Free PMC article. Review.
-
SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis.Front Cell Neurosci. 2015 Aug 25;9:336. doi: 10.3389/fncel.2015.00336. eCollection 2015. Front Cell Neurosci. 2015. PMID: 26379505 Free PMC article. Review.
-
Comprehensive Research on Past and Future Therapeutic Strategies Devoted to Treatment of Amyotrophic Lateral Sclerosis.Int J Mol Sci. 2022 Feb 22;23(5):2400. doi: 10.3390/ijms23052400. Int J Mol Sci. 2022. PMID: 35269543 Free PMC article. Review.
-
Riluzole But Not Melatonin Ameliorates Acute Motor Neuron Degeneration and Moderately Inhibits SOD1-Mediated Excitotoxicity Induced Disrupted Mitochondrial Ca2+ Signaling in Amyotrophic Lateral Sclerosis.Front Cell Neurosci. 2017 Jan 6;10:295. doi: 10.3389/fncel.2016.00295. eCollection 2016. Front Cell Neurosci. 2017. PMID: 28111541 Free PMC article.
-
Gut microbiome correlates with plasma lipids in amyotrophic lateral sclerosis.Brain. 2024 Feb 1;147(2):665-679. doi: 10.1093/brain/awad306. Brain. 2024. PMID: 37721161 Free PMC article.
References
-
- Andersen PM. Genetic aspects of amyotrophic lateral sclerosis/motor neuron disease. In: Shaw PJ, Strong MJ, editor. Motor neuron disorders. Philadelphia, PA, Butterworth Heinemann; 2003. pp. 207–235.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
