

Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes
- PMID: 18000544
- PMCID: PMC2048665
- DOI: 10.1371/journal.pone.0001172
Complexity reduction of polymorphic sequences (CRoPS): a novel approach for large-scale polymorphism discovery in complex genomes
Abstract
Application of single nucleotide polymorphisms (SNPs) is revolutionizing human bio-medical research. However, discovery of polymorphisms in low polymorphic species is still a challenging and costly endeavor, despite widespread availability of Sanger sequencing technology. We present CRoPS as a novel approach for polymorphism discovery by combining the power of reproducible genome complexity reduction of AFLP with Genome Sequencer (GS) 20/GS FLX next-generation sequencing technology. With CRoPS, hundreds-of-thousands of sequence reads derived from complexity-reduced genome sequences of two or more samples are processed and mined for SNPs using a fully-automated bioinformatics pipeline. We show that over 75% of putative maize SNPs discovered using CRoPS are successfully converted to SNPWave assays, confirming them to be true SNPs derived from unique (single-copy) genome sequences. By using CRoPS, polymorphism discovery will become affordable in organisms with high levels of repetitive DNA in the genome and/or low levels of polymorphism in the (breeding) germplasm without the need for prior sequence information.
Conflict of interest statement
Figures




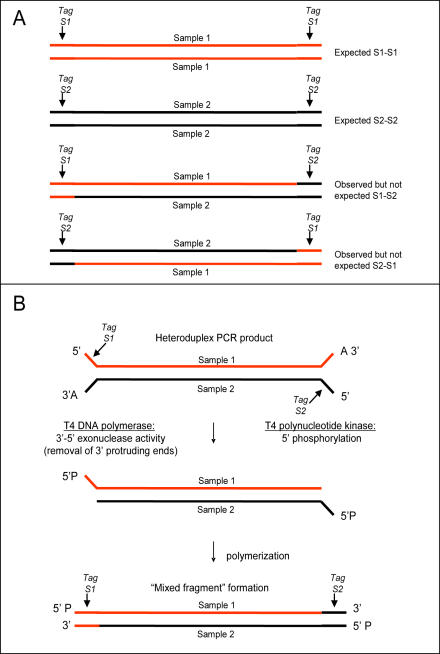
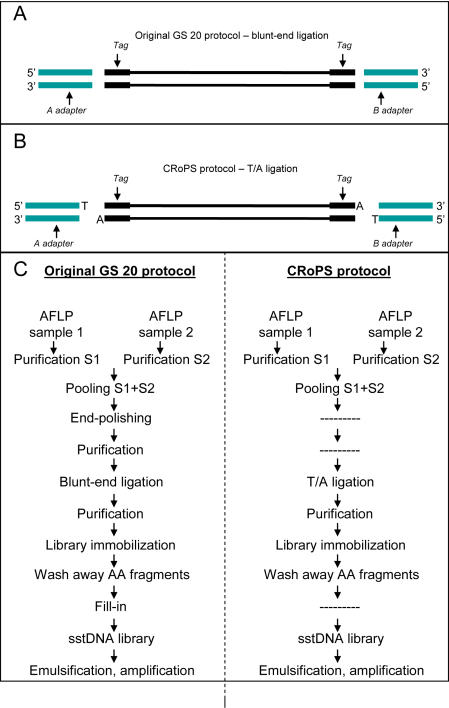
References
-
- The Arabidopsis Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
-
- International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793–800. - PubMed
-
- SanMiguel P, Tikhonov A, Jin Y-K, Motchoulskaia N, Zakharov D, et al. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274:765–768. - PubMed
-
- Li W, Zhang P, Fellers JP, Friebe B, Gill BS. Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 2004;40:500–511. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
