

Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana
- PMID: 18024567
- PMCID: PMC2174890
- DOI: 10.1105/tpc.107.055046
Genome-wide analysis of mRNA decay rates and their determinants in Arabidopsis thaliana
Abstract
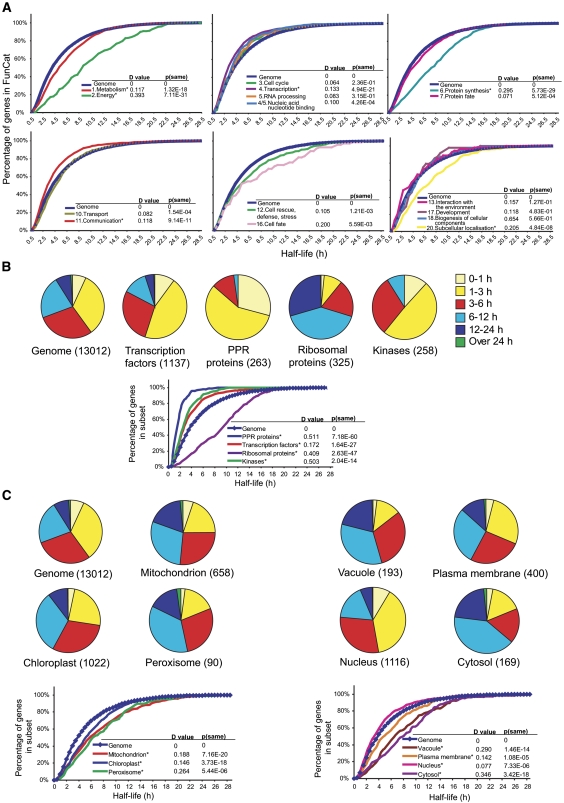
To gain a global view of mRNA decay in Arabidopsis thaliana, suspension cell cultures were treated with a transcriptional inhibitor, and microarrays were used to measure transcript abundance over time. The deduced mRNA half-lives varied widely, from minutes to >24 h. Three features of the transcript displayed a correlation with decay rates: (1) genes possessing at least one intron produce mRNA transcripts significantly more stable than those of intronless genes, and this was not related to overall length, sequence composition, or number of introns; (2) various sequence elements in the 3' untranslated region are enriched among short- and long-lived transcripts, and their multiple occurrence suggests combinatorial control of transcript decay; and (3) transcripts that are microRNA targets generally have short half-lives. The decay rate of transcripts correlated with subcellular localization and function of the encoded proteins. Analysis of transcript decay rates for genes encoding orthologous proteins between Arabidopsis, yeast, and humans indicated that yeast and humans had a higher percentage of transcripts with shorter half-lives and that the relative stability of transcripts from genes encoding proteins involved in cell cycle, transcription, translation, and energy metabolism is conserved. Comparison of decay rates with changes in transcript abundance under a variety of abiotic stresses reveal that a set of transcription factors are downregulated with similar kinetics to decay rates, suggesting that inhibition of their transcription is an important early response to abiotic stress.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
