

A code for transcription initiation in mammalian genomes
- PMID: 18032727
- PMCID: PMC2134772
- DOI: 10.1101/gr.6831208
A code for transcription initiation in mammalian genomes
Abstract
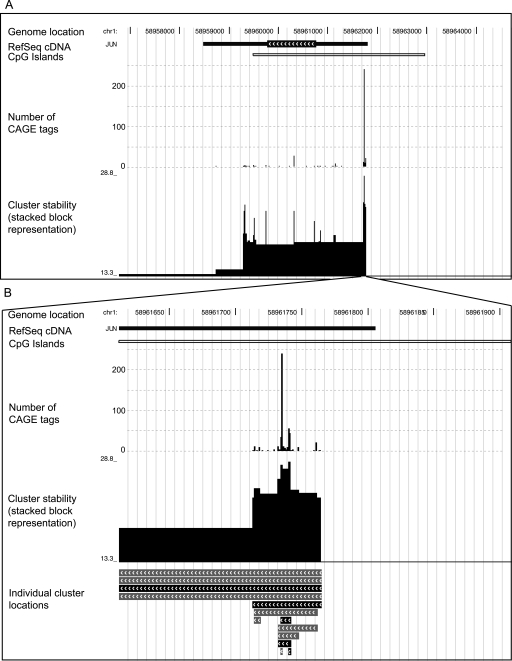
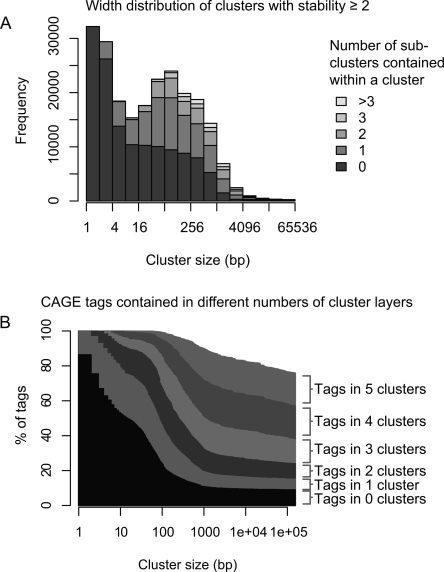
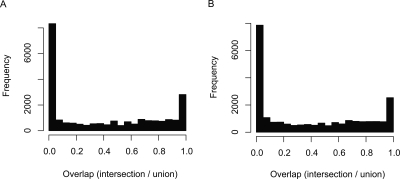
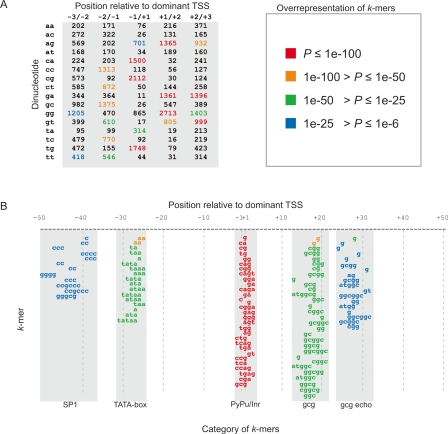
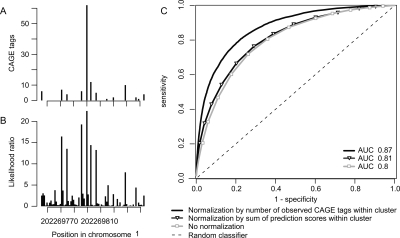
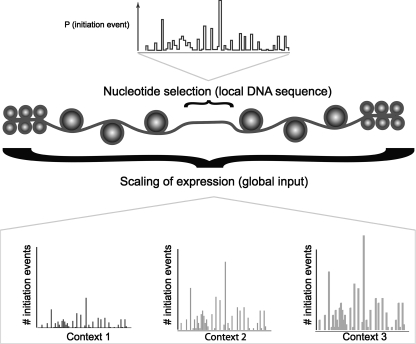
Genome-wide detection of transcription start sites (TSSs) has revealed that RNA Polymerase II transcription initiates at millions of positions in mammalian genomes. Most core promoters do not have a single TSS, but an array of closely located TSSs with different rates of initiation. As a rule, genes have more than one such core promoter; however, defining the boundaries between core promoters is not trivial. These discoveries prompt a re-evaluation of our models for transcription initiation. We describe a new framework for understanding the organization of transcription initiation. We show that initiation events are clustered on the chromosomes at multiple scales-clusters within clusters-indicating multiple regulatory processes. Within the smallest of such clusters, which can be interpreted as core promoters, the local DNA sequence predicts the relative transcription start usage of each nucleotide with a remarkable 91% accuracy, implying the existence of a DNA code that determines TSS selection. Conversely, the total expression strength of such clusters is only partially determined by the local DNA sequence. Thus, the overall control of transcription can be understood as a combination of large- and small-scale effects; the selection of transcription start sites is largely governed by the local DNA sequence, whereas the transcriptional activity of a locus is regulated at a different level; it is affected by distal features or events such as enhancers and chromatin remodeling.
Figures
References
-
- Akobeng A.K. Understanding diagnostic tests 3: Receiver operating characteristic curves. Acta Paediatr. 2007;96:644–647. - PubMed
-
- Bajic V.B., Tan S.L., Suzuki Y., Sugano S., Tan S.L., Suzuki Y., Sugano S., Suzuki Y., Sugano S., Sugano S. Promoter prediction analysis on the whole human genome. Nat. Biotechnol. 2004;22:1467–1473. - PubMed
-
- Bajic V.B., Brent M.R., Brown R.H., Frankish A., Harrow J., Ohler U., Solovyev V.V., Tan S.L., Brent M.R., Brown R.H., Frankish A., Harrow J., Ohler U., Solovyev V.V., Tan S.L., Brown R.H., Frankish A., Harrow J., Ohler U., Solovyev V.V., Tan S.L., Frankish A., Harrow J., Ohler U., Solovyev V.V., Tan S.L., Harrow J., Ohler U., Solovyev V.V., Tan S.L., Ohler U., Solovyev V.V., Tan S.L., Solovyev V.V., Tan S.L., Tan S.L. Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment. Genome Biol. 2006;7:S1–S3. doi: 10.1186/gb-2006-7-S1-S3. - DOI - PMC - PubMed
-
- Barrera L.O., Ren B., Ren B. The transcriptional regulatory code of eukaryotic cells - insights from genome-wide analysis of chromatin organization and transcription factor binding. Curr. Opin. Cell Biol. 2006;18:291–298. - PubMed
-
- Blake M.C., Jambou R.C., Swick A.G., Kahn J.W., Azizkhan J.C., Jambou R.C., Swick A.G., Kahn J.W., Azizkhan J.C., Swick A.G., Kahn J.W., Azizkhan J.C., Kahn J.W., Azizkhan J.C., Azizkhan J.C. Transcriptional initiation is controlled by upstream GC-box interactions in a TATAA-less promoter. Mol. Cell. Biol. 1990;10:6632–6641. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials