

TNF-alpha and adipocyte biology
- PMID: 18037376
- PMCID: PMC4304634
- DOI: 10.1016/j.febslet.2007.11.051
TNF-alpha and adipocyte biology
Abstract
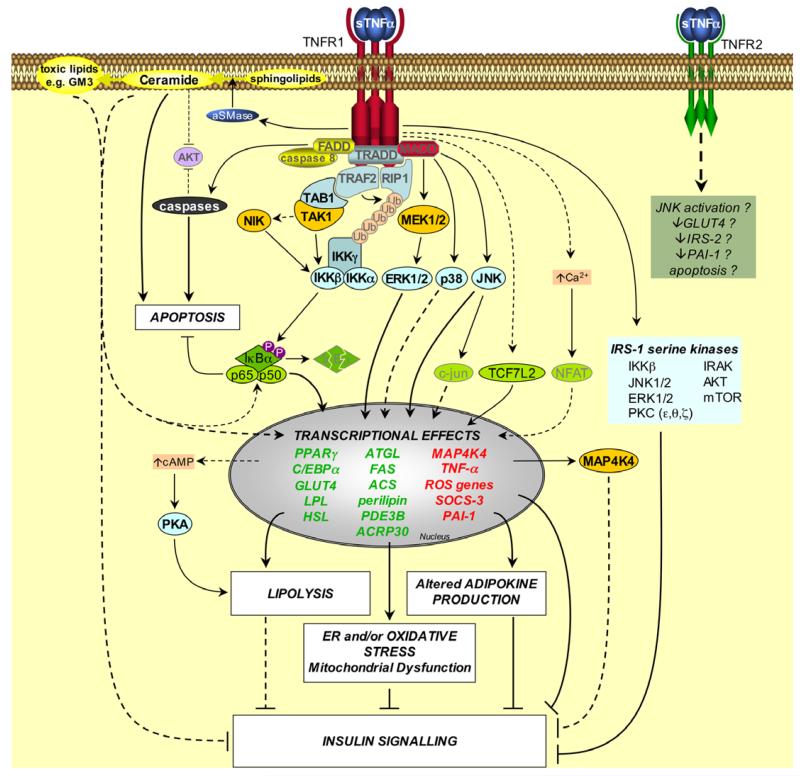
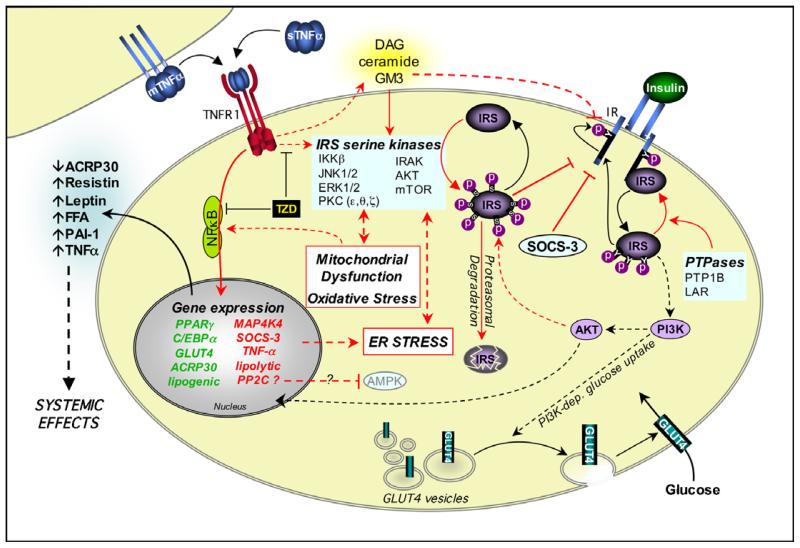
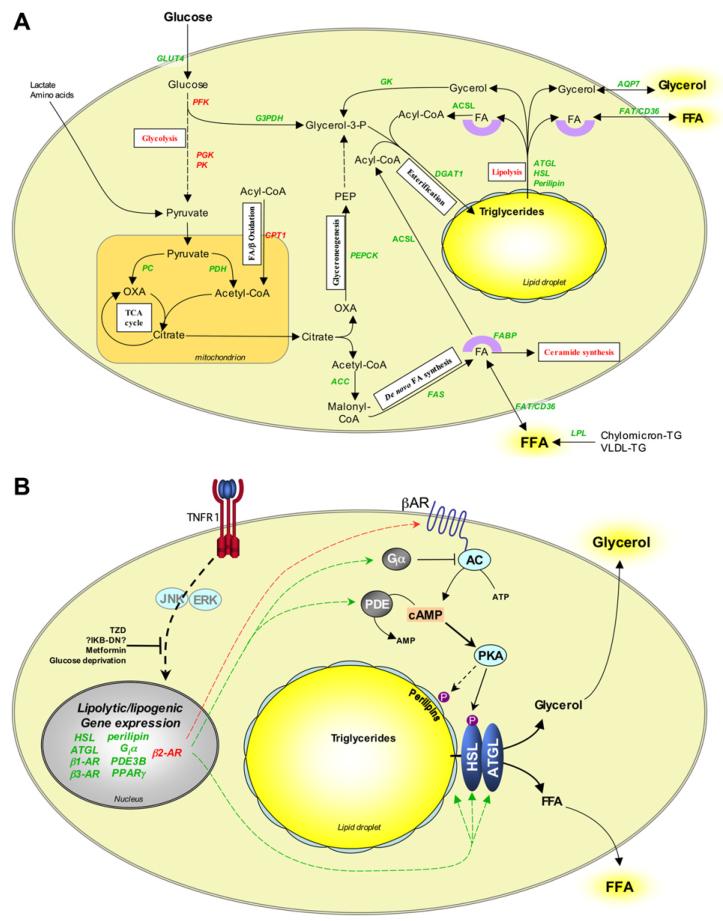
Dyslipidemia and insulin resistance are commonly associated with catabolic or lipodystrophic conditions (such as cancer and sepsis) and with pathological states of nutritional overload (such as obesity-related type 2 diabetes). Two common features of these metabolic disorders are adipose tissue dysfunction and elevated levels of tumour necrosis factor-alpha (TNF-alpha). Herein, we review the multiple actions of this pro-inflammatory adipokine on adipose tissue biology. These include inhibition of carbohydrate metabolism, lipogenesis, adipogenesis and thermogenesis and stimulation of lipolysis. TNF-alpha can also impact the endocrine functions of adipose tissue. Taken together, TNF-alpha contributes to metabolic dysregulation by impairing both adipose tissue function and its ability to store excess fuel. The molecular mechanisms that underlie these actions are discussed.
Figures
References
-
- Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:45–53. - PubMed
-
- Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. - PubMed
-
- Perez C, Albert I, DeFay K, Zachariades N, Gooding L, Kriegler M. A nonsecretable cell surface mutant of tumor necrosis factor (TNF) kills by cell-to-cell contact. Cell. 1990;63:251–258. - PubMed
-
- Xu H, Sethi JK, Hotamisligil GS. Transmembrane tumor necrosis factor (TNF)-alpha inhibits adipocyte differentiation by selectively activating TNF receptor 1. J. Biol. Chem. 1999;274:26287–26295. - PubMed
-
- Grell M. Tumor necrosis factor (TNF) receptors in cellular signaling of soluble and membrane-expressed TNF. J. Inflamm. 1995;47:8–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
