

A universal mechanism ties genotype to phenotype in trinucleotide diseases
- PMID: 18039028
- PMCID: PMC2082501
- DOI: 10.1371/journal.pcbi.0030235
A universal mechanism ties genotype to phenotype in trinucleotide diseases
Abstract
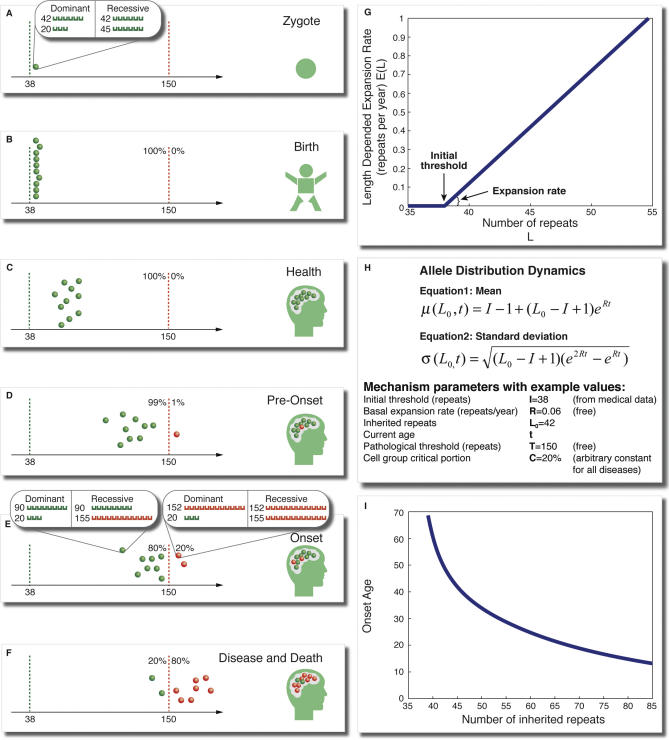
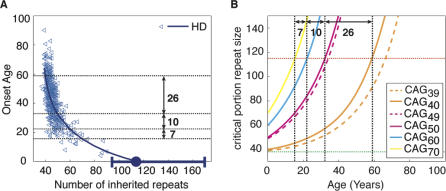
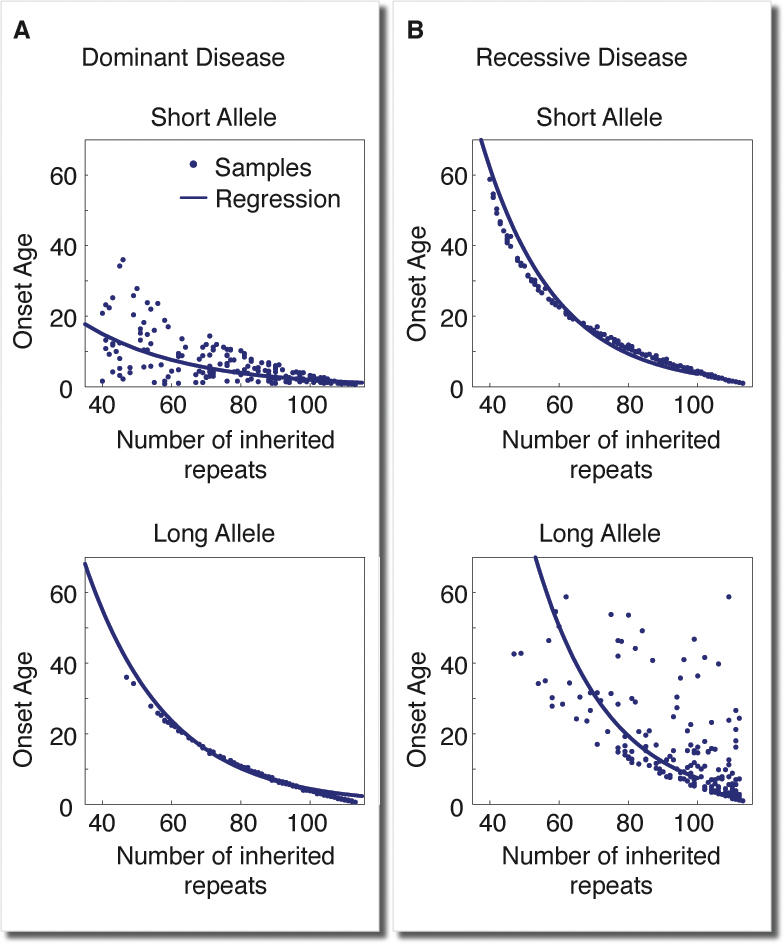
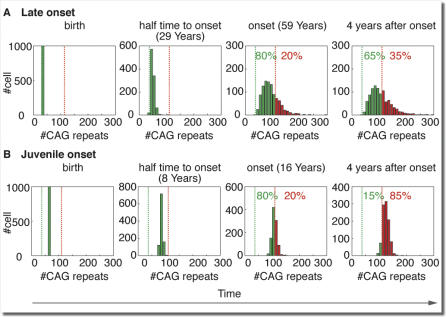
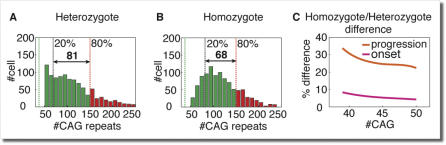
Trinucleotide hereditary diseases such as Huntington disease and Friedreich ataxia are cureless diseases associated with inheriting an abnormally large number of DNA trinucleotide repeats in a gene. The genes associated with different diseases are unrelated and harbor a trinucleotide repeat in different functional regions; therefore, it is striking that many of these diseases have similar correlations between their genotype, namely the number of inherited repeats and age of onset and progression phenotype. These correlations remain unexplained despite more than a decade of research. Although mechanisms have been proposed for several trinucleotide diseases, none of the proposals, being disease-specific, can account for the commonalities among these diseases. Here, we propose a universal mechanism in which length-dependent somatic repeat expansion occurs during the patient's lifetime toward a pathological threshold. Our mechanism uniformly explains for the first time to our knowledge the genotype-phenotype correlations common to trinucleotide disease and is well-supported by both experimental and clinical data. In addition, mathematical analysis of the mechanism provides simple explanations to a wide range of phenomena such as the exponential decrease of the age-of-onset curve, similar onset but faster progression in patients with Huntington disease with homozygous versus heterozygous mutation, and correlation of age of onset with length of the short allele but not with the long allele in Friedreich ataxia. If our proposed universal mechanism proves to be the core component of the actual mechanisms of specific trinucleotide diseases, it would open the search for a uniform treatment for all these diseases, possibly by delaying the somatic expansion process.
Conflict of interest statement
Figures
References
-
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. - PubMed
-
- Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: Mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–742. - PubMed
-
- Bates GP. History of genetic disease: The molecular genetics of Huntington disease—A history. Nat Rev Genet. 2005;6:766–773. - PubMed
-
- Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291–304. - PubMed
-
- Ranum LP, Day JW. Pathogenic RNA repeats: An expanding role in genetic disease. Trends Genet. 2004;20:506–512. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
