

Genetic variation and population structure in native Americans
- PMID: 18039031
- PMCID: PMC2082466
- DOI: 10.1371/journal.pgen.0030185
Genetic variation and population structure in native Americans
Abstract
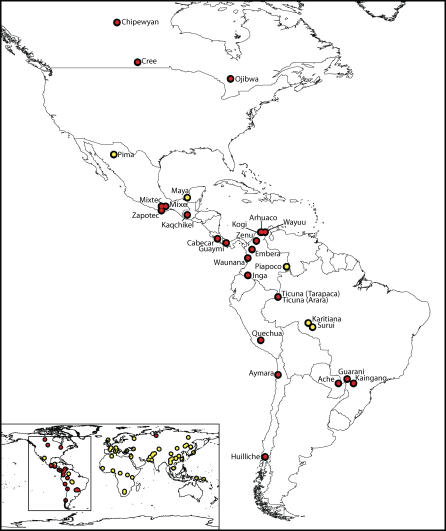
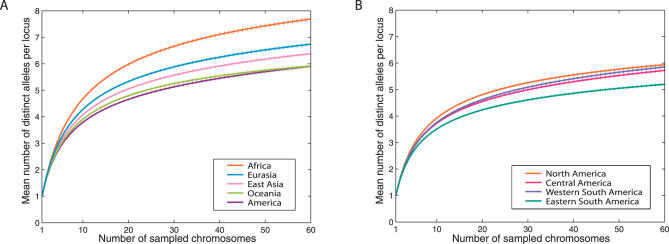
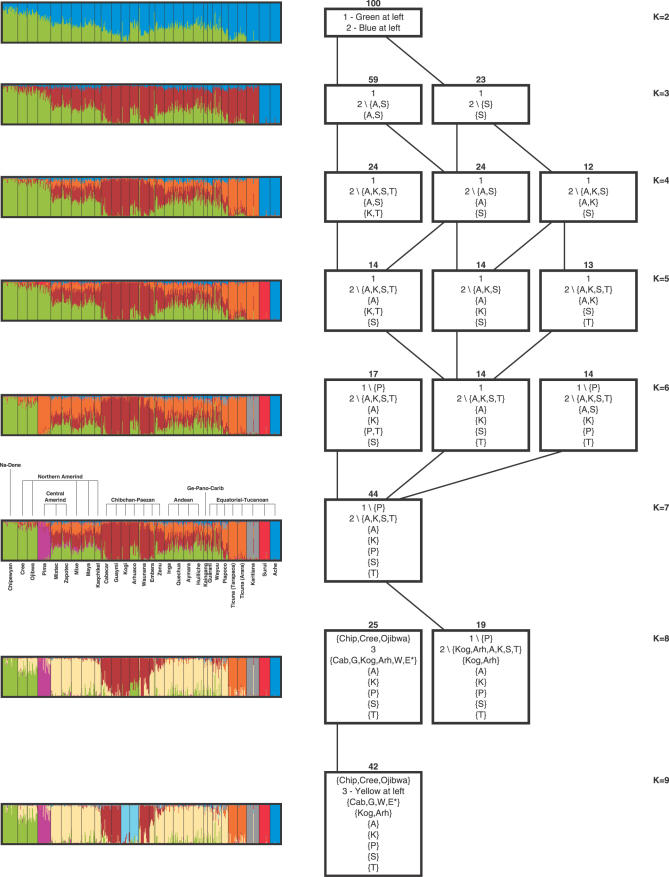
We examined genetic diversity and population structure in the American landmass using 678 autosomal microsatellite markers genotyped in 422 individuals representing 24 Native American populations sampled from North, Central, and South America. These data were analyzed jointly with similar data available in 54 other indigenous populations worldwide, including an additional five Native American groups. The Native American populations have lower genetic diversity and greater differentiation than populations from other continental regions. We observe gradients both of decreasing genetic diversity as a function of geographic distance from the Bering Strait and of decreasing genetic similarity to Siberians--signals of the southward dispersal of human populations from the northwestern tip of the Americas. We also observe evidence of: (1) a higher level of diversity and lower level of population structure in western South America compared to eastern South America, (2) a relative lack of differentiation between Mesoamerican and Andean populations, (3) a scenario in which coastal routes were easier for migrating peoples to traverse in comparison with inland routes, and (4) a partial agreement on a local scale between genetic similarity and the linguistic classification of populations. These findings offer new insights into the process of population dispersal and differentiation during the peopling of the Americas.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures







References
-
- Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton: Princeton University Press; 1994.
-
- Cavalli-Sforza LL, Feldman MW. The application of molecular genetic approaches to the study of human evolution. Nature Genet. 2003;33:S266–S275. - PubMed
-
- Jobling MA, Hurles ME, Tyler-Smith C. Human evolutionary genetics: origins, peoples & disease. New York: Garland Science; 2004.
-
- Tishkoff SA, Verrelli BC. Patterns of human genetic diversity: implications for human evolutionary history and disease. Annu Rev Genomics Hum Genet. 2003;4:293–340. - PubMed
-
- Di Rienzo A, Hudson RR. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 2005;21:596–601. - PubMed
