

Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo
- PMID: 18040046
- PMCID: PMC2148278
- DOI: 10.1073/pnas.0707952104
Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo
Abstract
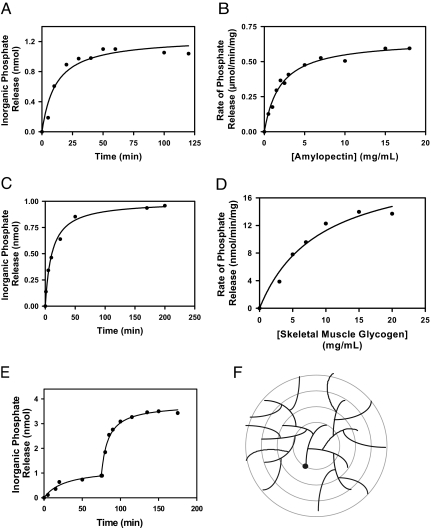
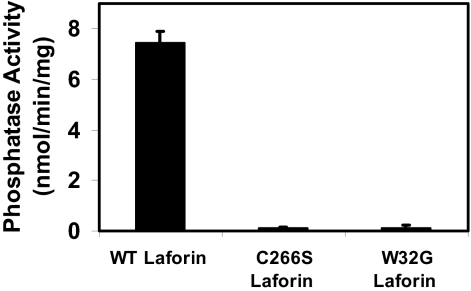
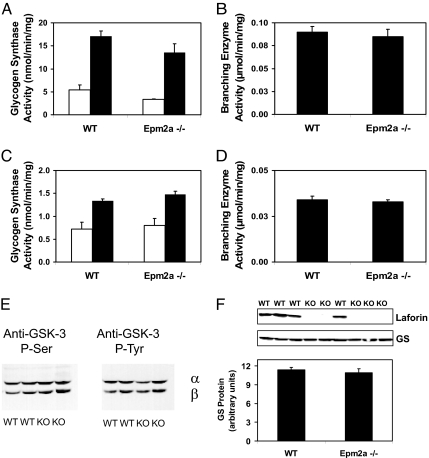
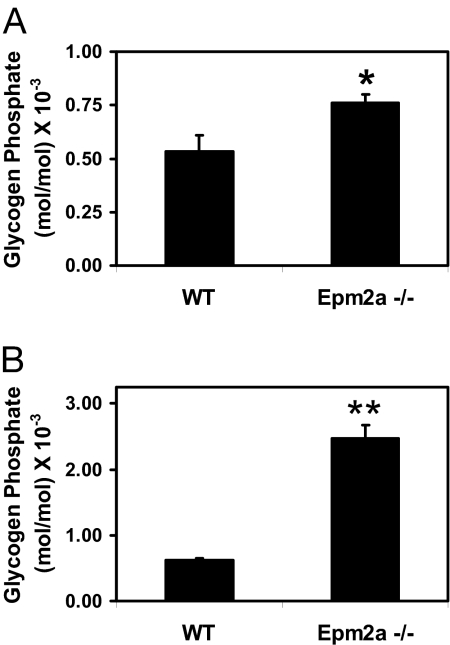
Lafora disease is a progressive myoclonus epilepsy with onset typically in the second decade of life and death within 10 years. Lafora bodies, deposits of abnormally branched, insoluble glycogen-like polymers, form in neurons, muscle, liver, and other tissues. Approximately half of the cases of Lafora disease result from mutations in the EPM2A gene, which encodes laforin, a member of the dual-specificity protein phosphatase family that additionally contains a glycogen binding domain. The molecular basis for the formation of Lafora bodies is completely unknown. Glycogen, a branched polymer of glucose, contains a small amount of covalently linked phosphate whose origin and function are obscure. We report here that recombinant laforin is able to release this phosphate in vitro, in a time-dependent reaction with an apparent K(m) for glycogen of 4.5 mg/ml. Mutations of laforin that disable the glycogen binding domain also eliminate its ability to dephosphorylate glycogen. We have also analyzed glycogen from a mouse model of Lafora disease, Epm2a(-/-) mice, which develop Lafora bodies in several tissues. Glycogen isolated from these mice had a 40% increase in the covalent phosphate content in liver and a 4-fold elevation in muscle. We propose that excessive phosphorylation of glycogen leads to aberrant branching and Lafora body formation. This study provides a molecular link between an observed biochemical property of laforin and the phenotype of a mouse model of Lafora disease. The results also have important implications for glycogen metabolism generally.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Abnormal metabolism of glycogen phosphate as a cause for Lafora disease.J Biol Chem. 2008 Dec 5;283(49):33816-25. doi: 10.1074/jbc.M807428200. Epub 2008 Oct 13. J Biol Chem. 2008. PMID: 18852261 Free PMC article.
-
The phosphatase activity of laforin is dispensable to rescue Epm2a-/- mice from Lafora disease.Brain. 2014 Mar;137(Pt 3):806-18. doi: 10.1093/brain/awt353. Epub 2014 Jan 14. Brain. 2014. PMID: 24430976
-
Increased laforin and laforin binding to glycogen underlie Lafora body formation in malin-deficient Lafora disease.J Biol Chem. 2012 Jul 20;287(30):25650-9. doi: 10.1074/jbc.M111.331611. Epub 2012 Jun 5. J Biol Chem. 2012. PMID: 22669944 Free PMC article.
-
Glycogen phosphorylation and Lafora disease.Mol Aspects Med. 2015 Dec;46:78-84. doi: 10.1016/j.mam.2015.08.003. Epub 2015 Aug 13. Mol Aspects Med. 2015. PMID: 26278984 Free PMC article. Review.
-
Are there errors in glycogen biosynthesis and is laforin a repair enzyme?FEBS Lett. 2011 Oct 20;585(20):3216-8. doi: 10.1016/j.febslet.2011.09.009. Epub 2011 Sep 16. FEBS Lett. 2011. PMID: 21930129 Free PMC article. Review.
Cited by
-
Laforin targets malin to glycogen in Lafora progressive myoclonus epilepsy.Dis Model Mech. 2023 Jan 1;16(1):dmm049802. doi: 10.1242/dmm.049802. Epub 2023 Jan 6. Dis Model Mech. 2023. PMID: 36511140 Free PMC article.
-
Molecular Architecture of the Inositol Phosphatase Siw14.Biochemistry. 2019 Feb 12;58(6):534-545. doi: 10.1021/acs.biochem.8b01044. Epub 2019 Jan 3. Biochemistry. 2019. PMID: 30548067 Free PMC article.
-
Lafora disease offers a unique window into neuronal glycogen metabolism.J Biol Chem. 2018 May 11;293(19):7117-7125. doi: 10.1074/jbc.R117.803064. Epub 2018 Feb 26. J Biol Chem. 2018. PMID: 29483193 Free PMC article. Review.
-
The dynamic life of the glycogen granule.J Biol Chem. 2018 May 11;293(19):7089-7098. doi: 10.1074/jbc.R117.802843. Epub 2018 Feb 26. J Biol Chem. 2018. PMID: 29483195 Free PMC article. Review.
-
Abnormal metabolism of glycogen phosphate as a cause for Lafora disease.J Biol Chem. 2008 Dec 5;283(49):33816-25. doi: 10.1074/jbc.M807428200. Epub 2008 Oct 13. J Biol Chem. 2008. PMID: 18852261 Free PMC article.
References
-
- Roach PJ. Curr Mol Med. 2002;2:101–120. - PubMed
-
- Cavanagh JB. Brain Res. 1999;29:265–295. - PubMed
-
- Wolfsdorf JI, Weinstein DA. Rev Endocrine Metabolic Disorders. 2003;4:95–102. - PubMed
-
- DiMauro S, Lamperti C. Muscle Nerve. 2001;24:984–999. - PubMed
-
- Chan EM, Andrade DM, Franceschetti S, Minassian B. Adv Neurol. 2005;95:47–57. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
