

Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function
- PMID: 18045994
- PMCID: PMC2230602
- DOI: 10.1091/mbc.e07-06-0587
Sequestration of mutated alpha1-antitrypsin into inclusion bodies is a cell-protective mechanism to maintain endoplasmic reticulum function
Abstract
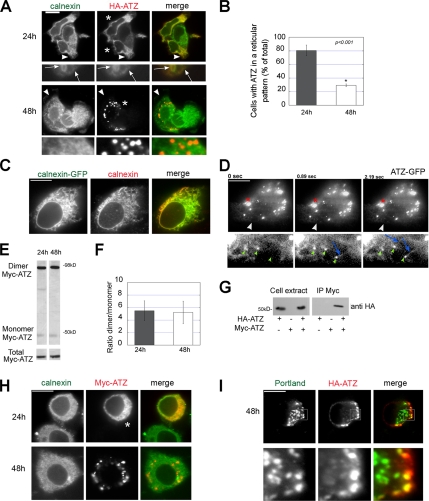
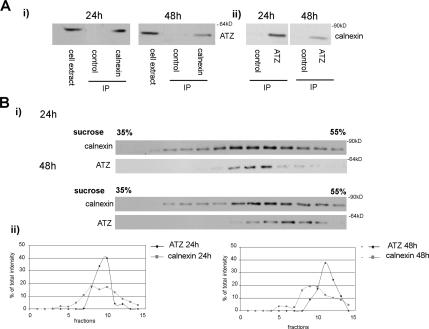
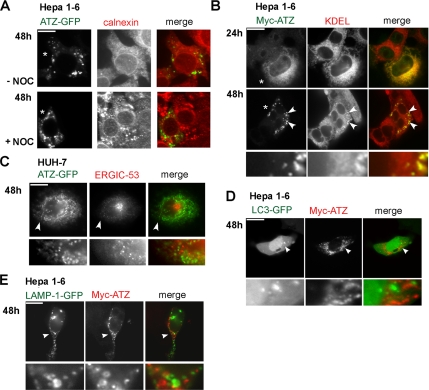
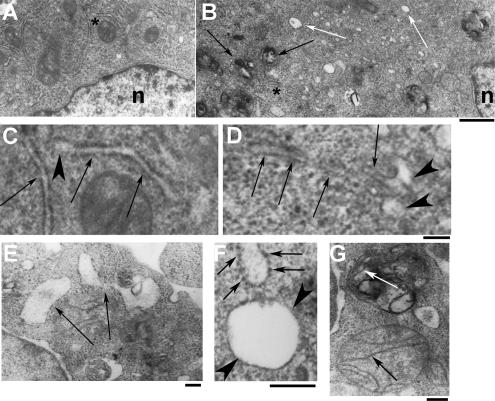
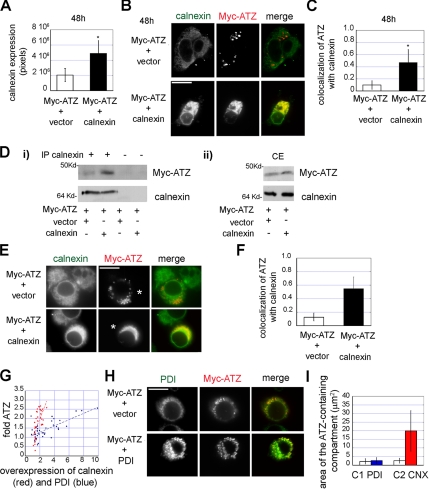
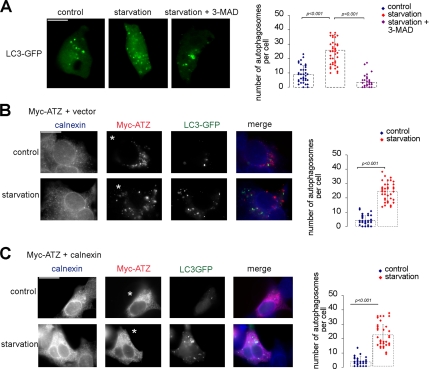
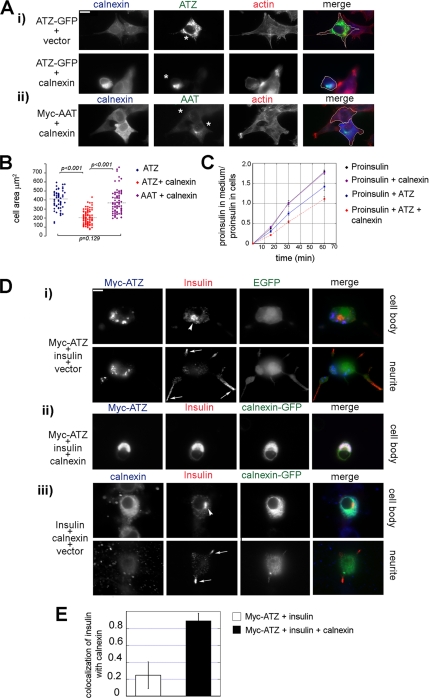
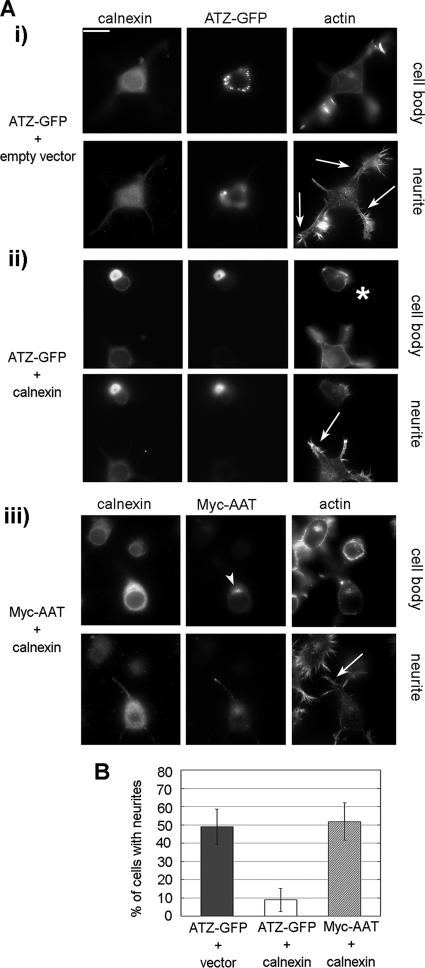
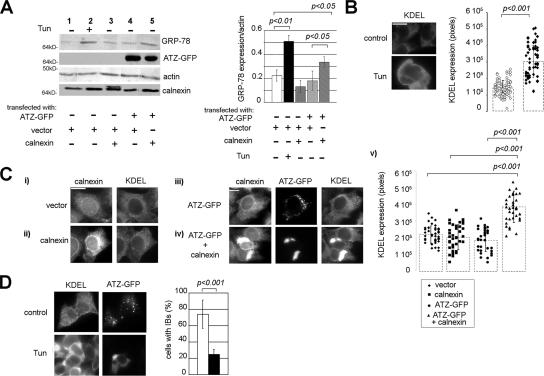
A variant alpha1-antitrypsin with E342K mutation has a high tendency to form intracellular polymers, and it is associated with liver disease. In the hepatocytes of individuals carrying the mutation, alpha1-antitrypsin localizes both to the endoplasmic reticulum (ER) and to membrane-surrounded inclusion bodies (IBs). It is unclear whether the IBs contribute to cell toxicity or whether they are protective to the cell. We found that in hepatoma cells, mutated alpha1-antitrypsin exited the ER and accumulated in IBs that were negative for autophagosomal and lysosomal markers, and contained several ER components, but not calnexin. Mutated alpha1-antitrypsin induced IBs also in neuroendocrine cells, showing that formation of these organelles is not cell type specific. In the presence of IBs, ER function was largely maintained. Increased levels of calnexin, but not of protein disulfide isomerase, inhibited formation of IBs and lead to retention of mutated alpha1-antitrypsin in the ER. In hepatoma cells, shift of mutated alpha1-antitrypsin localization to the ER by calnexin overexpression lead to cell shrinkage, ER stress, and impairment of the secretory pathway at the ER level. We conclude that segregation of mutated alpha1-antitrypsin from the ER to the IBs is a protective cell response to maintain a functional secretory pathway.
Figures
References
-
- Arrasate M., Mitra S., Schweitzer E. S., Segal M. R., Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. - PubMed
-
- Baldini G., Martelli A. M., Tabellini G., Horn C., Machaca K., Narducci P., Baldini G. Rabphilin localizes with the cell actin cytoskeleton and stimulates association of granules with F-actin cross-linked by α-actinin. J. Biol. Chem. 2005;280:34974–34984. - PubMed
-
- Bennett E. J., Bence N. F., Jayakumar R., Kopito R. R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. - PubMed
-
- Cabral C. M., Choudhury P., Liu Y., Sifers R. N. Processing by endoplasmic reticulum mannosidases partitions a secretion-impaired glycoprotein into distinct disposal pathways. J. Biol. Chem. 2000;275:25015–25022. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
