

Glucolipotoxicity: fuel excess and beta-cell dysfunction
- PMID: 18048763
- PMCID: PMC2528858
- DOI: 10.1210/er.2007-0023
Glucolipotoxicity: fuel excess and beta-cell dysfunction
Abstract
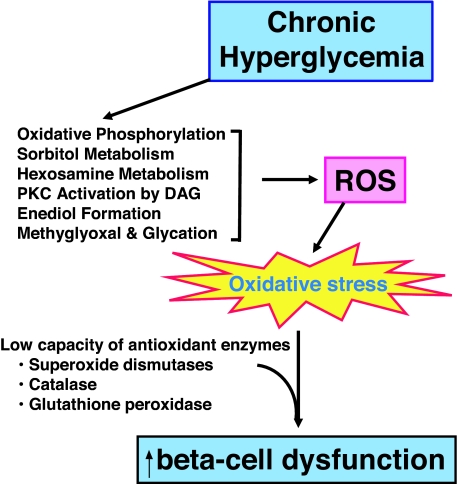
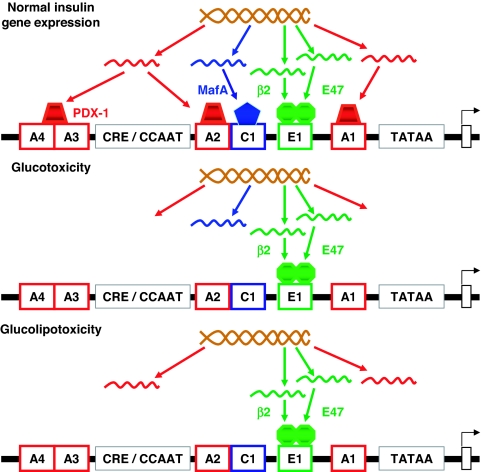
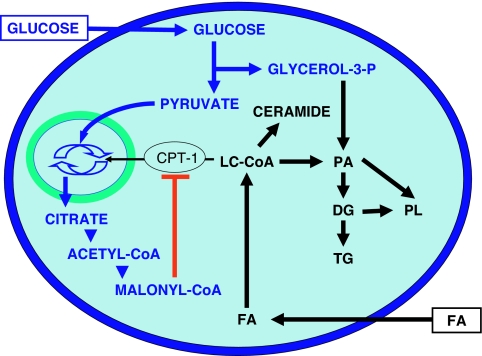
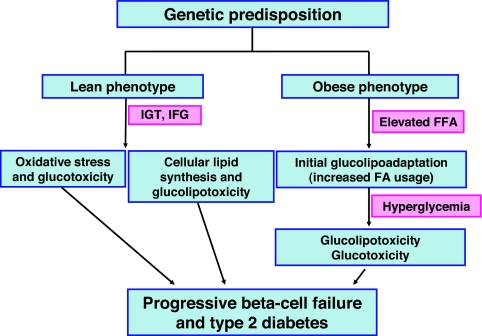
Glucotoxicity, lipotoxicity, and glucolipotoxicity are secondary phenomena that are proposed to play a role in all forms of type 2 diabetes. The underlying concept is that once the primary pathogenesis of diabetes is established, probably involving both genetic and environmental forces, hyperglycemia and very commonly hyperlipidemia ensue and thereafter exert additional damaging or toxic effects on the beta-cell. In addition to their contribution to the deterioration of beta-cell function after the onset of the disease, elevations of plasma fatty acid levels that often accompany insulin resistance may, as glucose levels begin to rise outside of the normal range, also play a pathogenic role in the early stages of the disease. Because hyperglycemia is a prerequisite for lipotoxicity to occur, the term glucolipotoxicity, rather than lipotoxicity, is more appropriate to describe deleterious effects of lipids on beta-cell function. In vitro and in vivo evidence supporting the concept of glucotoxicity is presented first, as well as a description of the underlying mechanisms with an emphasis on the role of oxidative stress. Second, we discuss the functional manifestations of glucolipotoxicity on insulin secretion, insulin gene expression, and beta-cell death, and the role of glucose in the mechanisms of glucolipotoxicity. Finally, we attempt to define the role of these phenomena in the natural history of beta-cell compensation, decompensation, and failure during the course of type 2 diabetes.
Figures
References
-
- DeFronzo RA 1999 Pathogenesis of type 2 diabetes: metabolic and molecular implications for identifying diabetes genes. Diabetes Rev 5:177–269
-
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P 2007 A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885 - PubMed
-
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S 2007 Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336 - PubMed
-
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M 2007 A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
