

CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures
- PMID: 18052539
- PMCID: PMC2098860
- DOI: 10.1371/journal.pcbi.0030232
CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures
Abstract
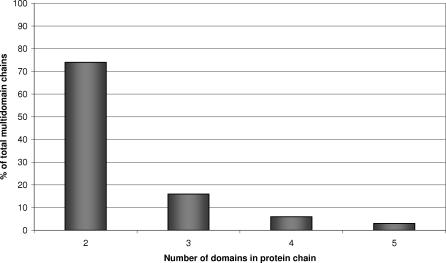
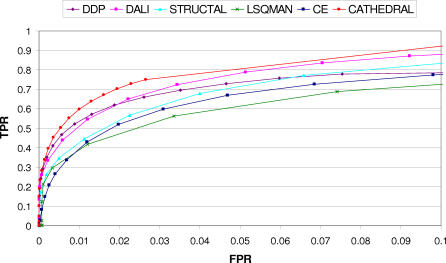
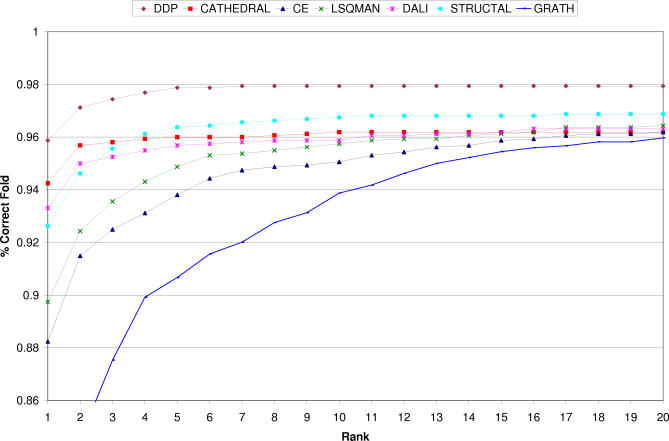
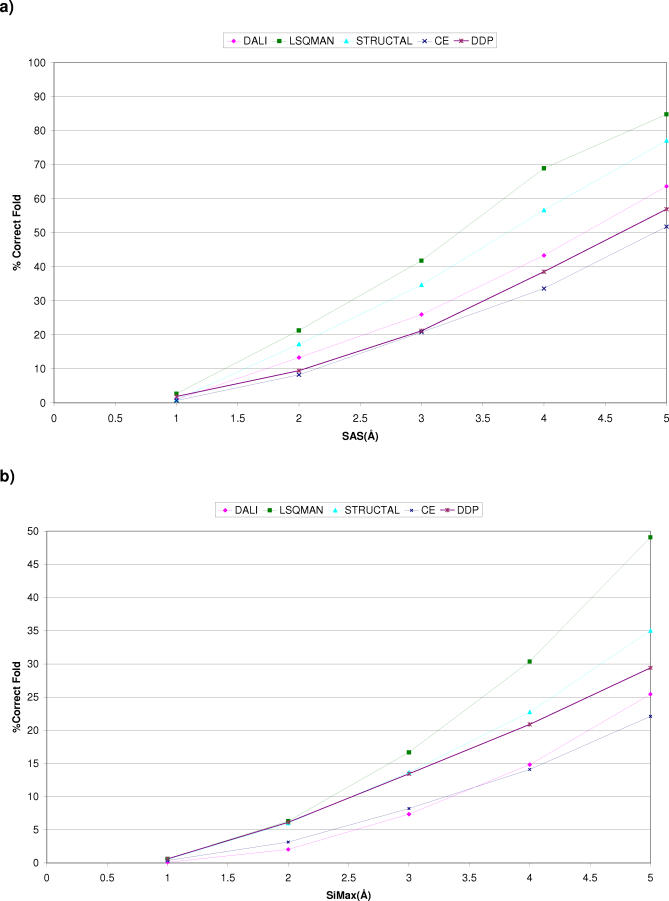
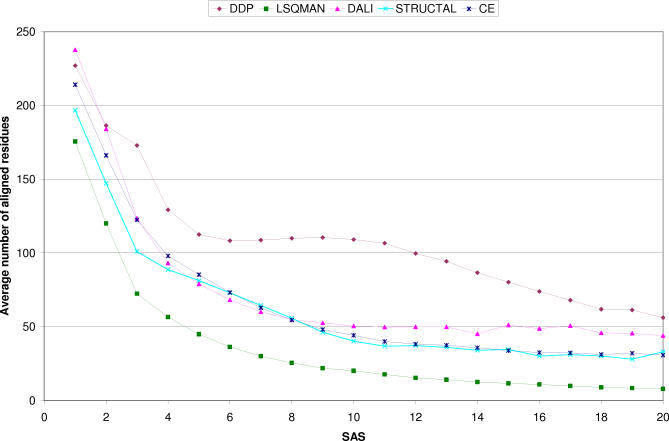
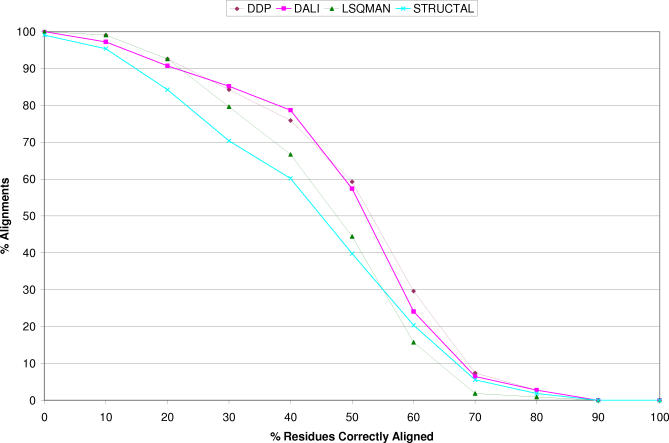
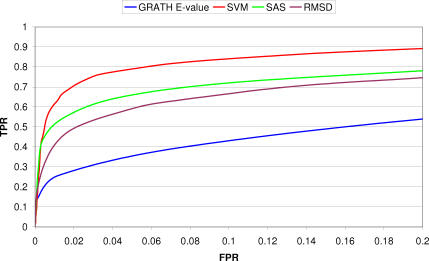
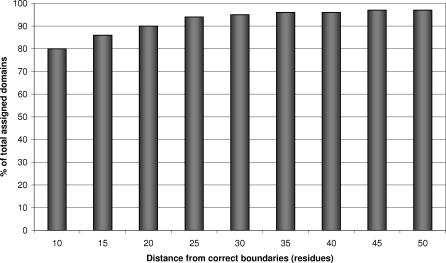
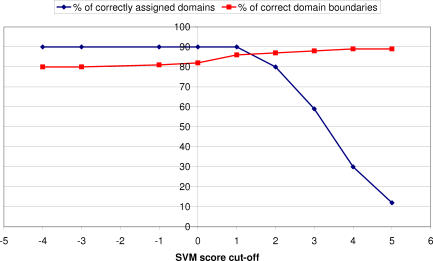
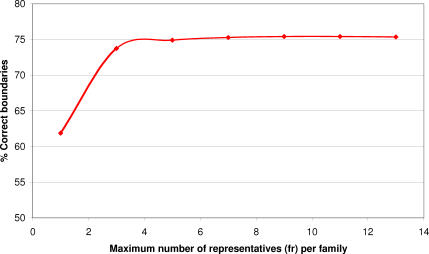
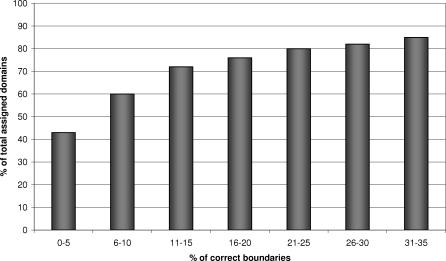
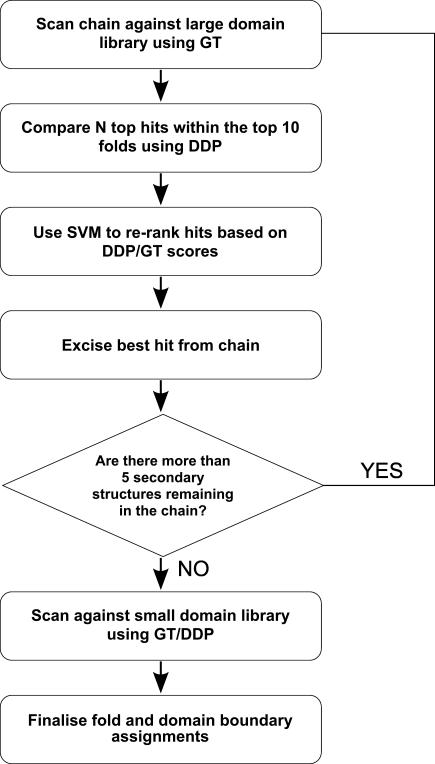
We present CATHEDRAL, an iterative protocol for determining the location of previously observed protein folds in novel multidomain protein structures. CATHEDRAL builds on the features of a fast secondary-structure-based method (using graph theory) to locate known folds within a multidomain context and a residue-based, double-dynamic programming algorithm, which is used to align members of the target fold groups against the query protein structure to identify the closest relative and assign domain boundaries. To increase the fidelity of the assignments, a support vector machine is used to provide an optimal scoring scheme. Once a domain is verified, it is excised, and the search protocol is repeated in an iterative fashion until all recognisable domains have been identified. We have performed an initial benchmark of CATHEDRAL against other publicly available structure comparison methods using a consensus dataset of domains derived from the CATH and SCOP domain classifications. CATHEDRAL shows superior performance in fold recognition and alignment accuracy when compared with many equivalent methods. If a novel multidomain structure contains a known fold, CATHEDRAL will locate it in 90% of cases, with <1% false positives. For nearly 80% of assigned domains in a manually validated test set, the boundaries were correctly delineated within a tolerance of ten residues. For the remaining cases, previously classified domains were very remotely related to the query chain so that embellishments to the core of the fold caused significant differences in domain sizes and manual refinement of the boundaries was necessary. To put this performance in context, a well-established sequence method based on hidden Markov models was only able to detect 65% of domains, with 33% of the subsequent boundaries assigned within ten residues. Since, on average, 50% of newly determined protein structures contain more than one domain unit, and typically 90% or more of these domains are already classified in CATH, CATHEDRAL will considerably facilitate the automation of protein structure classification.
Conflict of interest statement
Figures
References
-
- Apic G, Gough J, Teichmann SA. Domain combinations in archaeal, eubacterial and eukaryotic proteomes. J Mol Biol. 2001;310:311–325. - PubMed
-
- Orengo CA, Jones DT, Thornton JM. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. - PubMed
-
- Coulson AF, Moult J. A unifold, mesofold, and superfold model of protein fold use. Proteins. 2002;46:61–71. - PubMed
-
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
