

Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors
- PMID: 18054559
- PMCID: PMC2180834
- DOI: 10.1053/j.gastro.2007.09.008
Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors
Abstract
Background & aims: Chronic inflammation is a risk factor for colon cancer in patients with ulcerative colitis (UC). The molecular mechanisms linking inflammation and colon carcinogenesis are incompletely understood. We tested the hypothesis that Toll-like receptor 4 (TLR4) is involved in tumorigenesis in the setting of chronic inflammation.
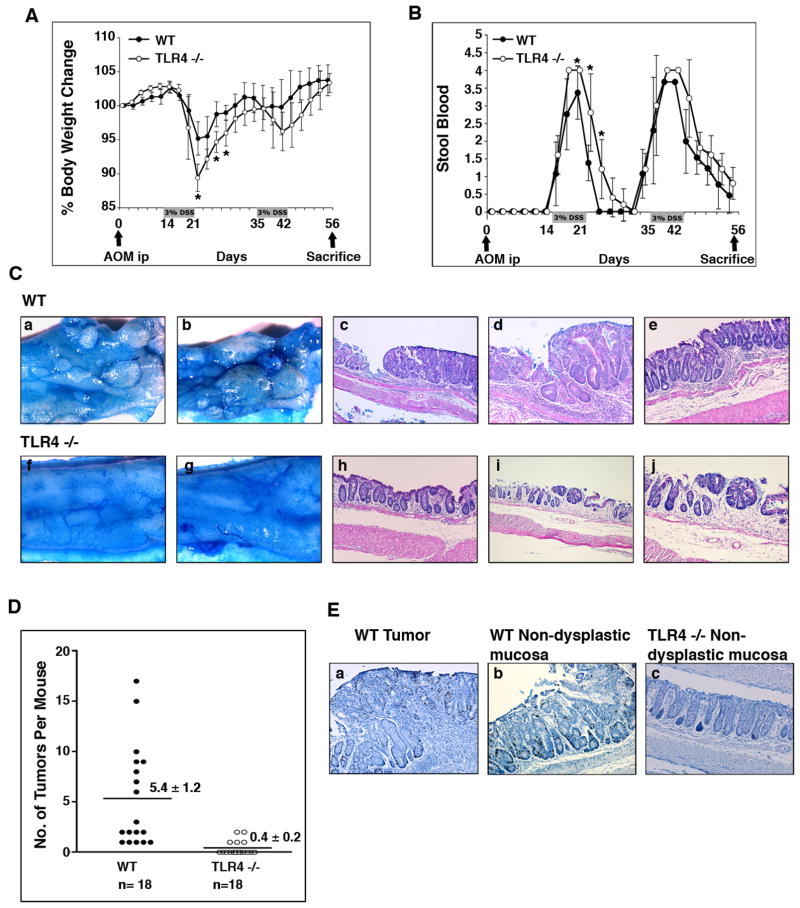
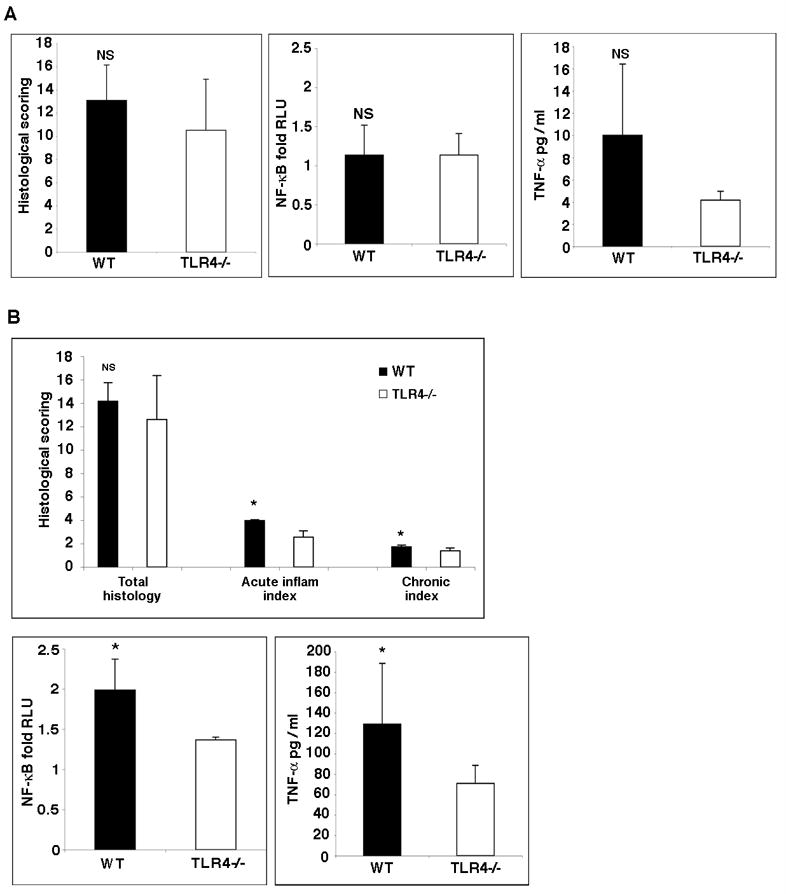
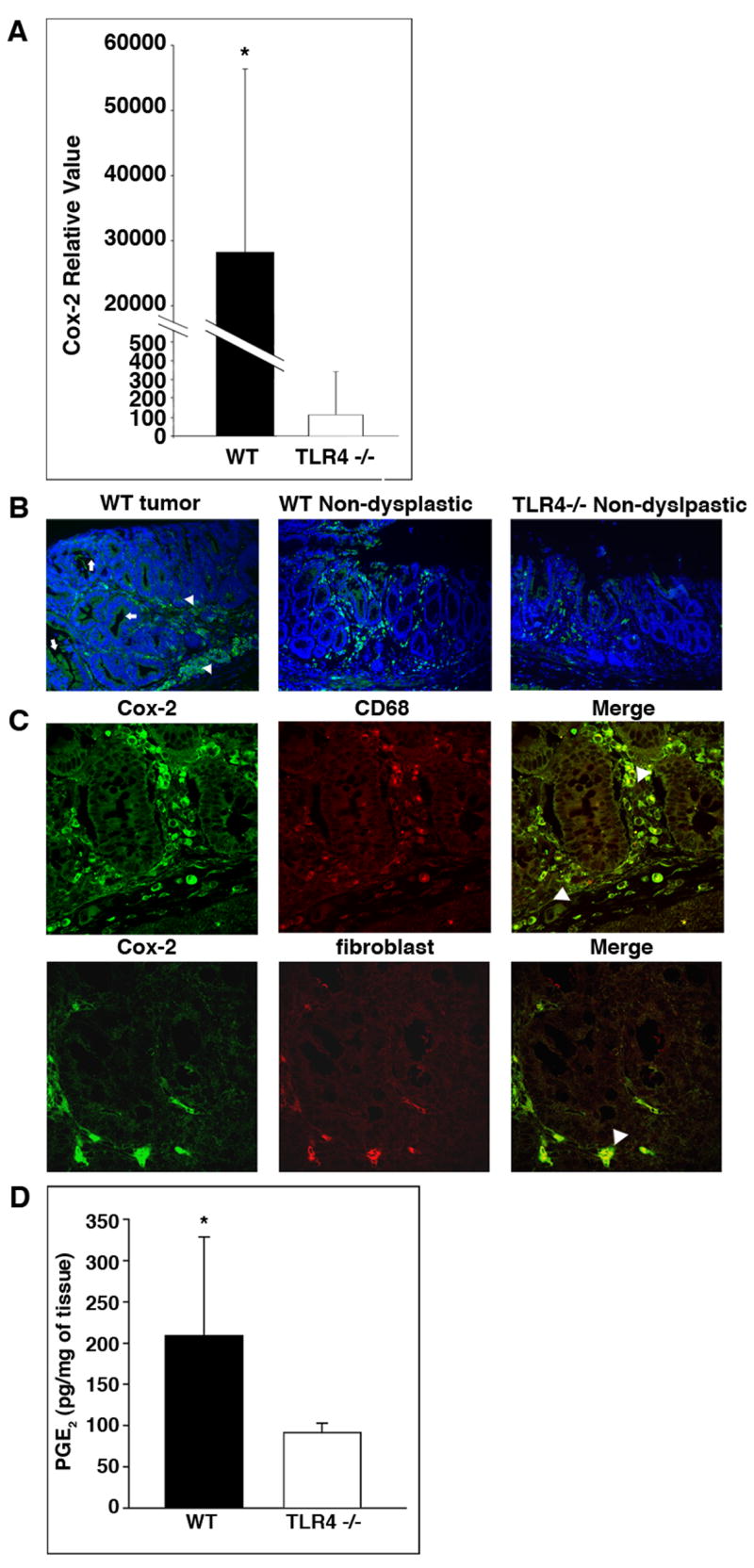
Methods: Tissues from UC patients with cancer were examined for TLR4 expression. Colitis-associated neoplasia was induced using azoxymethane injection followed by dextran sodium sulfate treatment in TLR4-deficient or wild-type mice. Inflammation, polyps, and microscopic dysplasia were scored. Cyclooxygenase (Cox)-2 and prostaglandin E(2) production were analyzed by real-time polymerase chain reaction, immunohistochemistry, or enzyme immunoassay. Epidermal growth factor receptor (EGFR) phosphorylation and amphiregulin production were examined by Western blot analysis and enzyme-linked immunosorbent assay, respectively.
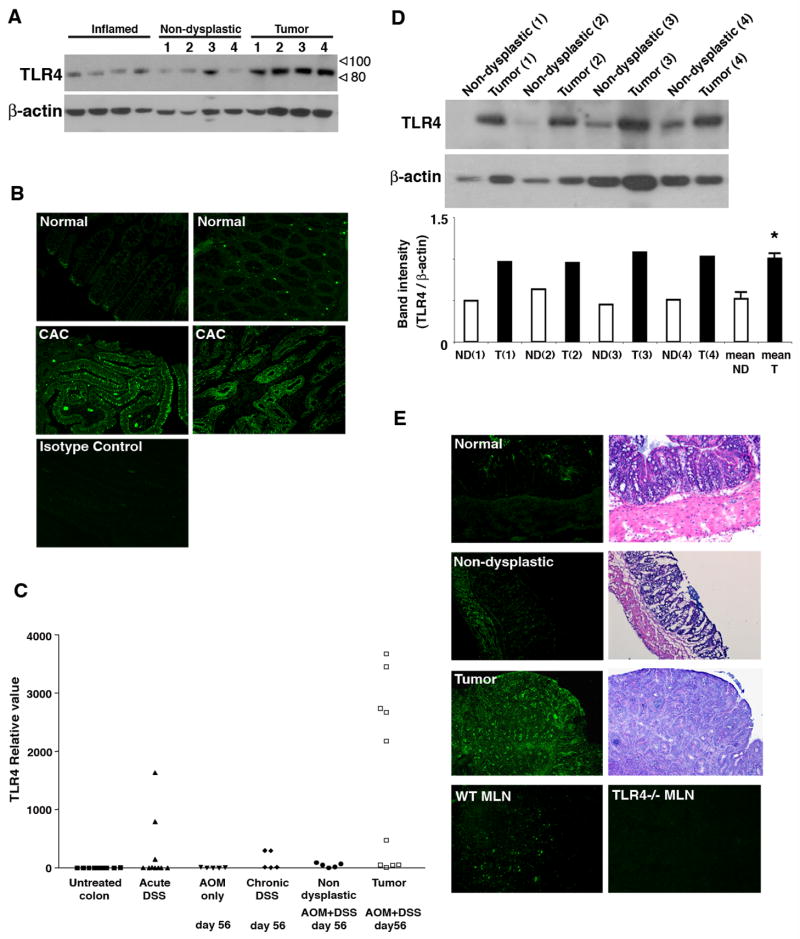
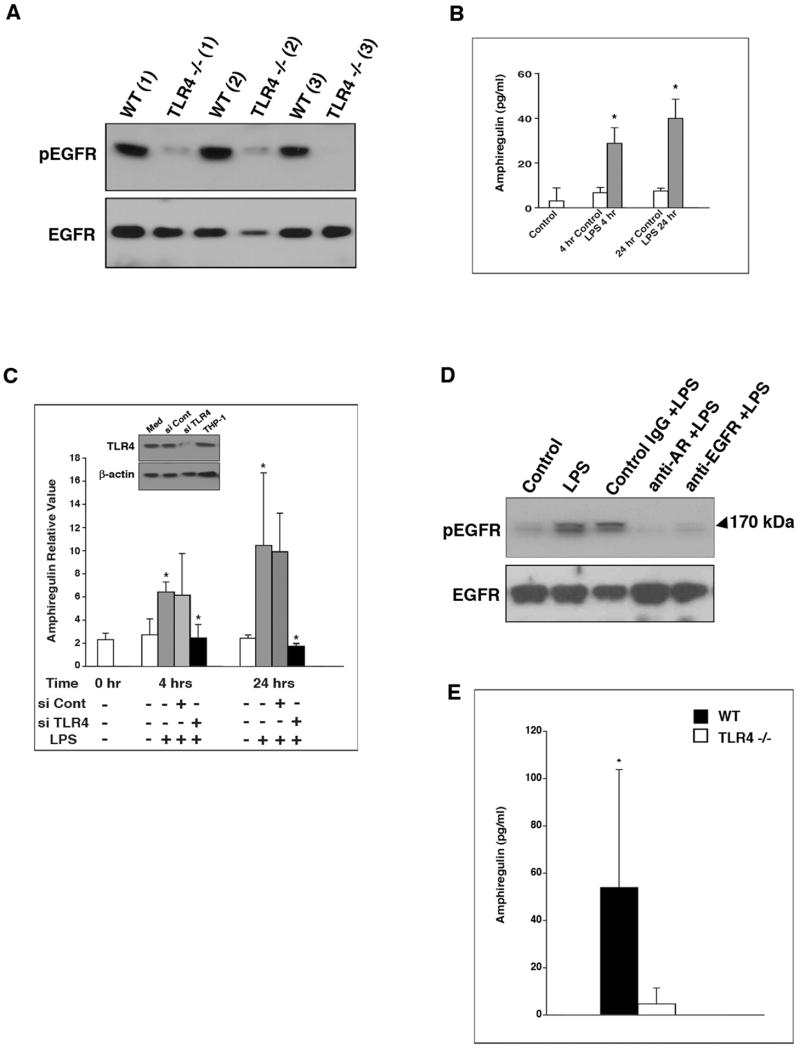
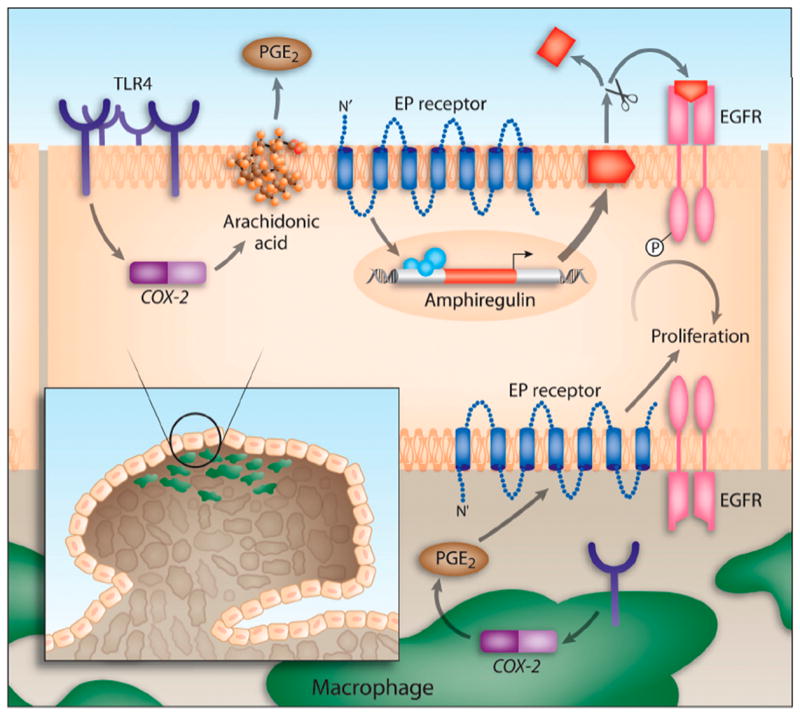
Results: We show that TLR4 is overexpressed in human and murine inflammation-associated colorectal neoplasia. TLR4-deficient mice were protected markedly from colon carcinogenesis. Mechanistically, we show that TLR4 is responsible for induction of Cox-2, increased prostaglandin E(2) production, and activation of EGFR signaling in chronic colitis. Amphiregulin, an EGFR ligand, was induced in a TLR4, Cox-2-dependent fashion and contributes to activation of EGFR phosphorylation in colonic epithelial cells.
Conclusions: TLR4 signaling is critical for colon carcinogenesis in chronic colitis. TLR4 activation appears to promote the development of colitis-associated cancer by mechanisms including enhanced Cox-2 expression and increased EGFR signaling. Inhibiting TLR4 signaling may be useful in the prevention or treatment of colitis-associated cancer.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
References
-
- Rao VP, Poutahidis T, Ge Z, Nambiar PR, Boussahmain C, Wang YY, Horwitz BH, Fox JG, Erdman SE. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006;66:7395–400. - PubMed
-
- Gunter MJ, Stolzenberg-Solomon R, Cross AJ, Leitzmann MF, Weinstein S, Wood RJ, Virtamo J, Taylor PR, Albanes D, Sinha R. A prospective study of serum C-reactive protein and colorectal cancer risk in men. Cancer Res. 2006;66:2483–7. - PubMed
-
- Surveillance, Epidemiology, and End Results (SEER) Program and the National Center for Health Statistics. http://seer.cancer.gov/
-
- Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
