

Probing protein fold space with a simplified model
- PMID: 18054792
- PMCID: PMC2254652
- DOI: 10.1016/j.jmb.2007.10.087
Probing protein fold space with a simplified model
Abstract
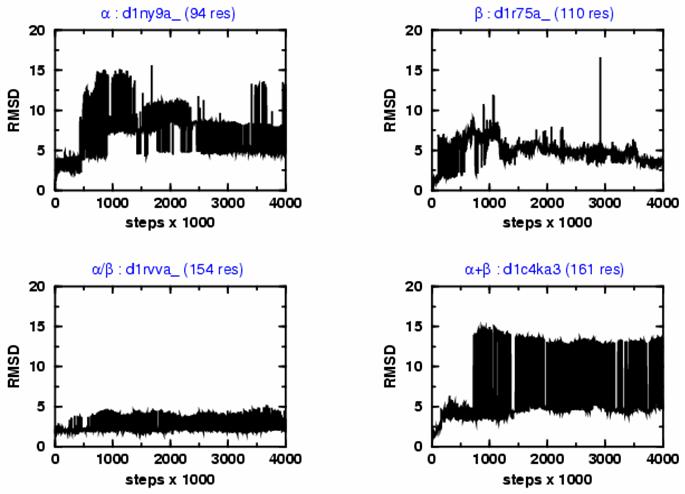
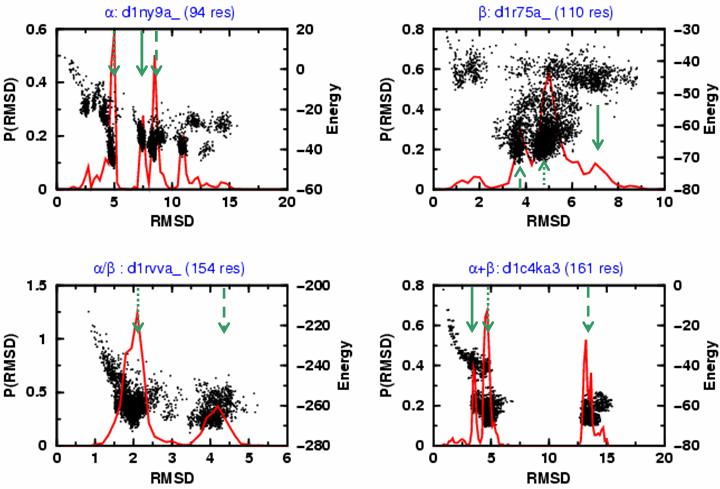
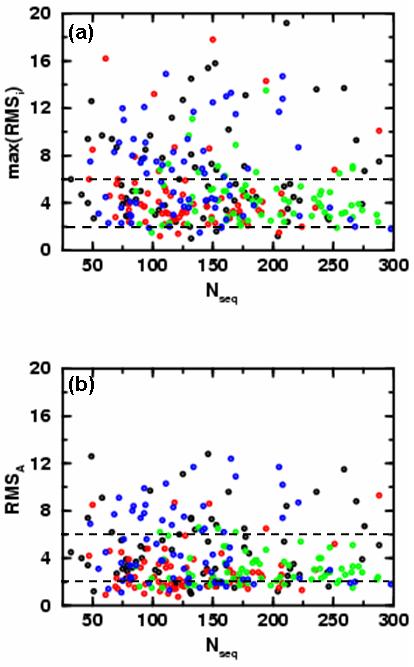
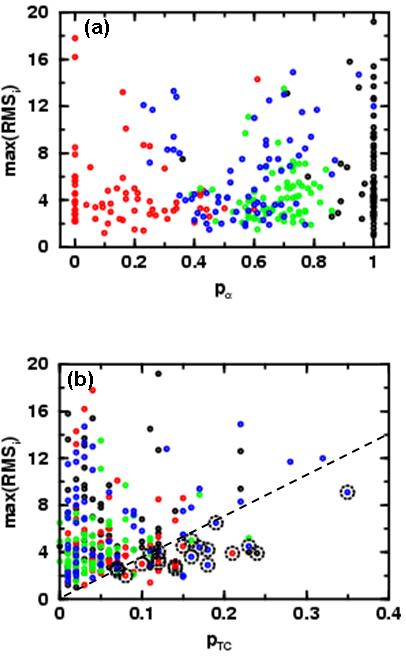
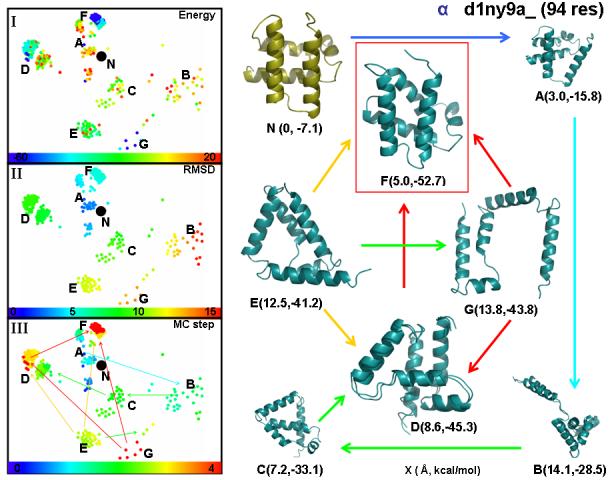
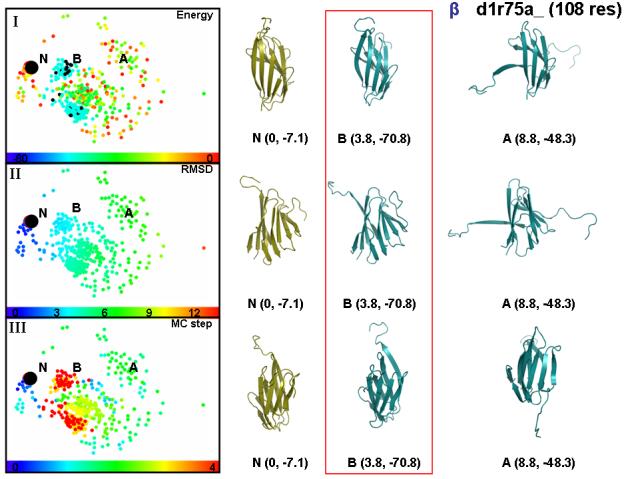
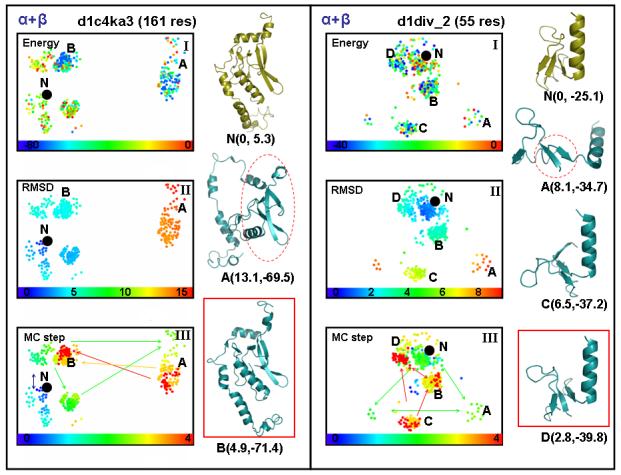
We probe the stability and near-native energy landscape of protein fold space using powerful conformational sampling methods together with simple reduced models and statistical potentials. Fold space is represented by a set of 280 protein domains spanning all topological classes and having a wide range of lengths (33-300 residues) amino acid composition and number of secondary structural elements. The degrees of freedom are taken as the loop torsion angles. This choice preserves the native secondary structure but allows the tertiary structure to change. The proteins are represented by three-point per residue, three-dimensional models with statistical potentials derived from a knowledge-based study of known protein structures. When this space is sampled by a combination of parallel tempering and equi-energy Monte Carlo, we find that the three-point model captures the known stability of protein native structures with stable energy basins that are near-native (all alpha: 4.77 A, all beta: 2.93 A, alpha/beta: 3.09 A, alpha+beta: 4.89 A on average and within 6 A for 71.41%, 92.85%, 94.29% and 64.28% for all-alpha, all-beta, alpha/beta and alpha+beta, classes, respectively). Denatured structures also occur and these have interesting structural properties that shed light on the different landscape characteristics of alpha and beta folds. We find that alpha/beta proteins with alternating alpha and beta segments (such as the beta-barrel) are more stable than proteins in other fold classes.
Figures
Similar articles
-
Reduced alphabet of prebiotic amino acids optimally encodes the conformational space of diverse extant protein folds.BMC Evol Biol. 2019 Jul 30;19(1):158. doi: 10.1186/s12862-019-1464-6. BMC Evol Biol. 2019. PMID: 31362700 Free PMC article.
-
TOUCHSTONE II: a new approach to ab initio protein structure prediction.Biophys J. 2003 Aug;85(2):1145-64. doi: 10.1016/S0006-3495(03)74551-2. Biophys J. 2003. PMID: 12885659 Free PMC article.
-
De novo backbone and sequence design of an idealized alpha/beta-barrel protein: evidence of stable tertiary structure.J Mol Biol. 2003 Jan 3;325(1):163-74. doi: 10.1016/s0022-2836(02)01206-8. J Mol Biol. 2003. PMID: 12473459
-
Dynamic Monte Carlo simulations of a new lattice model of globular protein folding, structure and dynamics.J Mol Biol. 1991 Sep 20;221(2):499-531. doi: 10.1016/0022-2836(91)80070-b. J Mol Biol. 1991. PMID: 1920430 Review.
-
Folding protein alpha-carbon chains into compact forms by Monte Carlo methods.Proteins. 1992 Nov;14(3):409-20. doi: 10.1002/prot.340140310. Proteins. 1992. PMID: 1438179 Review.
Cited by
-
Global view of the protein universe.Proc Natl Acad Sci U S A. 2014 Aug 12;111(32):11691-6. doi: 10.1073/pnas.1403395111. Epub 2014 Jul 28. Proc Natl Acad Sci U S A. 2014. PMID: 25071170 Free PMC article.
-
Nonlinearities in protein space limit the utility of informatics in protein biophysics.Proteins. 2015 Nov;83(11):1923-8. doi: 10.1002/prot.24916. Epub 2015 Sep 10. Proteins. 2015. PMID: 26315852 Free PMC article.
-
Evolution of tunnels in α/β-hydrolase fold proteins-What can we learn from studying epoxide hydrolases?PLoS Comput Biol. 2022 May 17;18(5):e1010119. doi: 10.1371/journal.pcbi.1010119. eCollection 2022 May. PLoS Comput Biol. 2022. PMID: 35580137 Free PMC article.
-
Splitting statistical potentials into meaningful scoring functions: testing the prediction of near-native structures from decoy conformations.BMC Struct Biol. 2009 Nov 16;9:71. doi: 10.1186/1472-6807-9-71. BMC Struct Biol. 2009. PMID: 19917096 Free PMC article.
-
A review of visualisations of protein fold networks and their relationship with sequence and function.Biol Rev Camb Philos Soc. 2023 Feb;98(1):243-262. doi: 10.1111/brv.12905. Epub 2022 Oct 9. Biol Rev Camb Philos Soc. 2023. PMID: 36210328 Free PMC article. Review.
References
-
- Ponting C, Russell R. The natural history of protein domains. Annu. Rev. Biophys. Biomol. Struct. 2002;31:45–71. - PubMed
-
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP:a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. - PubMed
-
- Orengo CA, Michie AD, Jones S, Jones DT, Swindels MB, Thornton JM. CATH: a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. - PubMed
-
- Holm L, Sander C. Mapping the Protein Universe. Science. 1996;273:595–602. - PubMed
-
- Levitt M, Chothia C. Structural Patterns in Globular Proteins. Nature. 1976;261:552–558. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
