

Inhibiting protease-activated receptor 4 limits myocardial ischemia/reperfusion injury in rat hearts by unmasking adenosine signaling
- PMID: 18055876
- PMCID: PMC2935083
- DOI: 10.1124/jpet.107.133595
Inhibiting protease-activated receptor 4 limits myocardial ischemia/reperfusion injury in rat hearts by unmasking adenosine signaling
Abstract
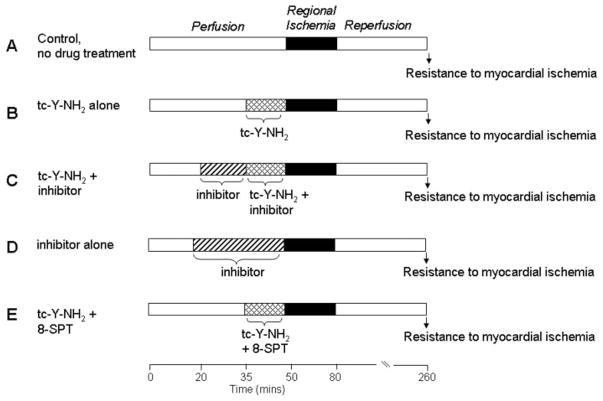
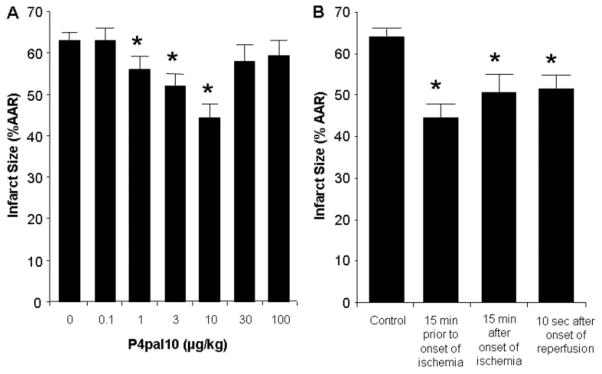
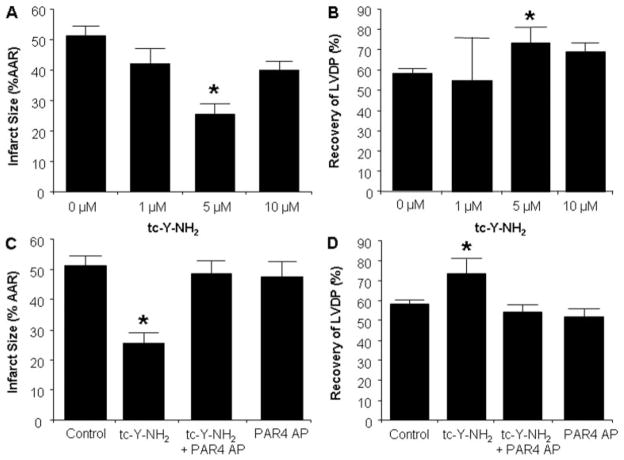
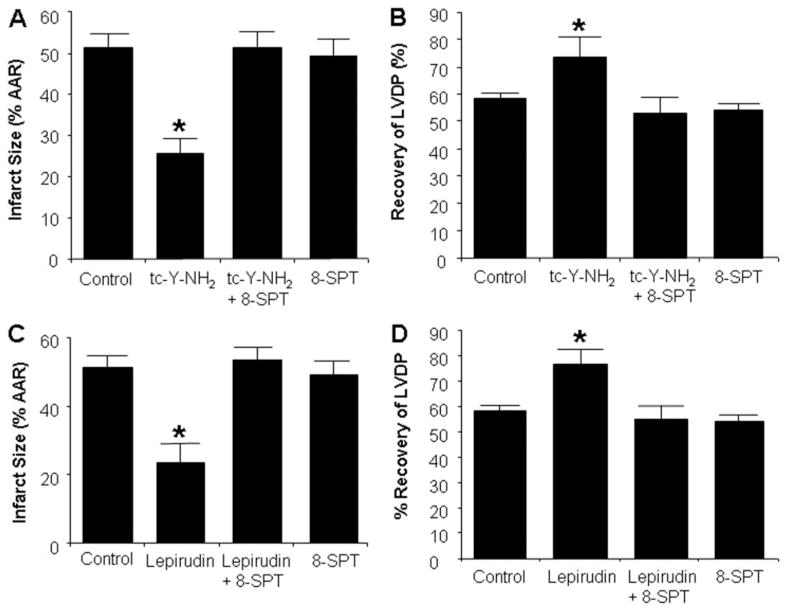
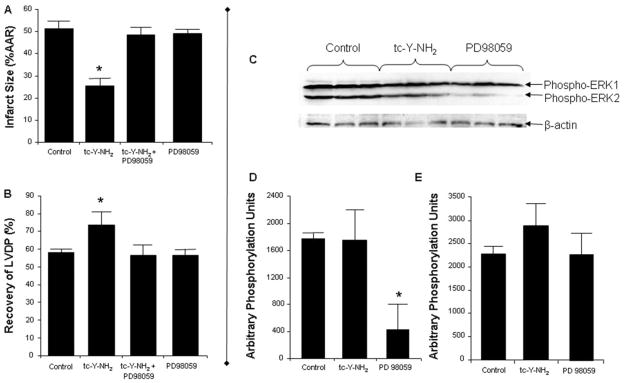
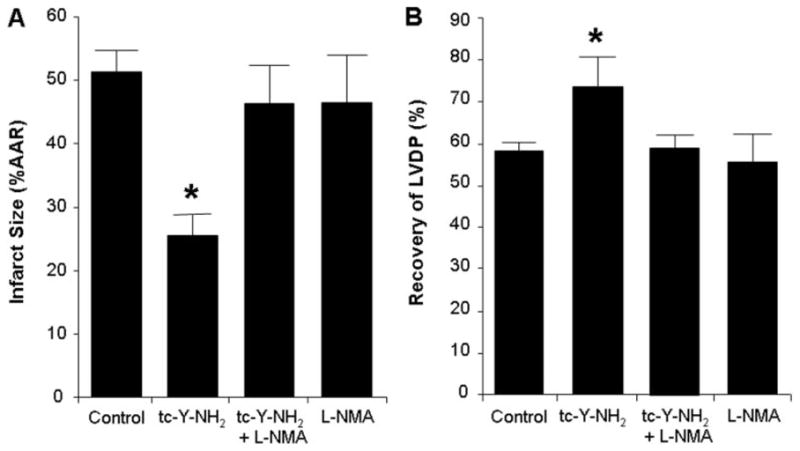
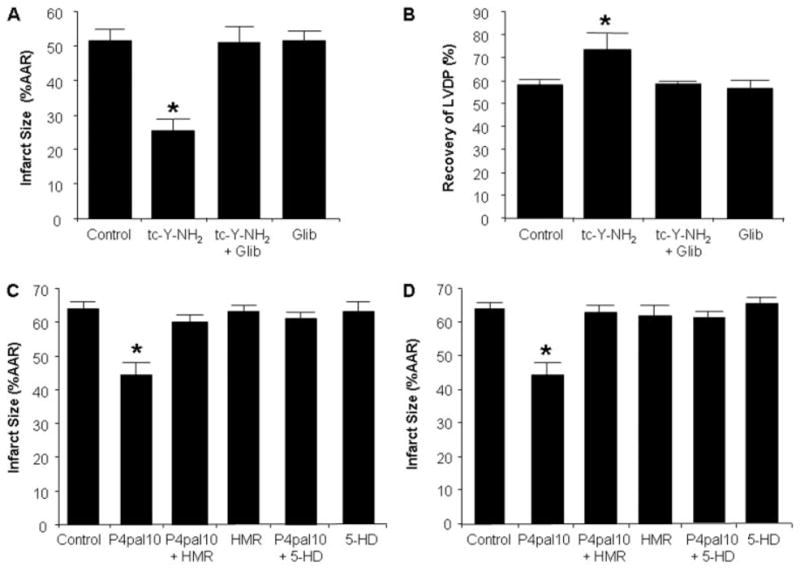
Harnessing endogenous cardioprotectants is a novel therapeutic strategy to combat ischemia/reperfusion (I/R) injury. Thrombin causes I/R injury, whereas exogenous adenosine prevents I/R injury. We hypothesized that blocking thrombin receptor activation with a protease-activated receptor (PAR) 4 antagonist would unmask the cardioprotective effects of endogenous adenosine. The protective role of two structurally unrelated PAR4 antagonists, trans-cinnamoyl-YPGKF-amide (tc-Y-NH(2)) and palmitoyl-SGRRYGHALR-amide (P4pal10), were evaluated in two rat models of myocardial I/R injury. P4pal10 (10 microg/kg) treatment before ischemia significantly decreased infarct size (IS) by 31, 21, and 19% when given before, during, and after ischemia in the in vivo model. tc-Y-NH(2) (5 microM) treatment before ischemia decreased IS by 51% in the in vitro model and increased recovery of ventricular function by 26%. To assess whether the cardioprotective effects of PAR4 blockade were due to endogenous adenosine, isolated hearts were treated with a nonselective adenosine receptor blocker, 8-sulfaphenyltheophylline (8-SPT), and tc-Y-NH(2) before ischemia. 8-SPT abolished the protective effects of tc-Y-NH(2) but did not affect IS when given alone. Adenosine-mediated survival pathways were then explored. The cardioprotective effects of tc-Y-NH(2) were abolished by inhibition of Akt (wortmannin), extracellular signal-regulated kinase 1/2 [PD98059 (2'-amino-3'-methoxyflavone)], nitric-oxide synthase [N(G)-monomethyl-l-arginine (l-NMA)], and K(ATP) channels (glibenclamide). PD98059, l-NMA, and glibenclamide alone had no effect on cardioprotection in vitro. Furthermore, inhibition of mitochondrial K(ATP) channels [5-hydroxydecanoic acid (5-HD)] and sarcolemmal K(ATP) channels (sodium (5-(2-(5-chloro-2-methoxybenzamido)ethyl)-2-methoxyphenylsulfonyl)(methylcarbamothioyl)amide; HMR 1098) abolished P4pal10-induced cardioprotection in vivo. Thrombin receptor blockade by PAR4 inhibition provides protection against injury from myocardial I/R by unmasking adenosine receptor signaling and supports the hypothesis of a coupling between thrombin receptors and adenosine receptors.
Figures

Similar articles
-
SCH 79797, a selective PAR1 antagonist, limits myocardial ischemia/reperfusion injury in rat hearts.Basic Res Cardiol. 2007 Jul;102(4):350-8. doi: 10.1007/s00395-007-0653-4. Epub 2007 Apr 30. Basic Res Cardiol. 2007. PMID: 17468933 Free PMC article.
-
Parstatin(1-26): the putative signal peptide of protease-activated receptor 1 confers potent protection from myocardial ischemia-reperfusion injury.J Pharmacol Exp Ther. 2010 Mar;332(3):898-905. doi: 10.1124/jpet.109.162602. Epub 2009 Dec 15. J Pharmacol Exp Ther. 2010. PMID: 20008957
-
Parstatin: a cryptic peptide involved in cardioprotection after ischaemia and reperfusion injury.Cardiovasc Res. 2009 Jul 15;83(2):325-34. doi: 10.1093/cvr/cvp122. Epub 2009 Apr 20. Cardiovasc Res. 2009. PMID: 19380418 Free PMC article.
-
GSK3beta inhibition and K(ATP) channel opening mediate acute opioid-induced cardioprotection at reperfusion.Basic Res Cardiol. 2007 Jul;102(4):341-9. doi: 10.1007/s00395-007-0651-6. Epub 2007 Apr 23. Basic Res Cardiol. 2007. PMID: 17450314
-
Adenosine-A1 receptors activation restores the suppressed cardioprotective effects of ischemic preconditioning in hyperhomocysteinemic rat hearts.J Cardiovasc Pharmacol. 2009 Sep;54(3):204-12. doi: 10.1097/FJC.0b013e3181b04cc5. J Cardiovasc Pharmacol. 2009. PMID: 19568176
Cited by
-
Polymorphism in the protease-activated receptor-4 gene region associates with platelet activation and perioperative myocardial injury.Am J Hematol. 2012 Feb;87(2):161-6. doi: 10.1002/ajh.22244. Epub 2012 Jan 7. Am J Hematol. 2012. PMID: 22228373 Free PMC article.
-
Platelets Are at the Nexus of Vascular Diseases.Front Cardiovasc Med. 2019 Sep 11;6:132. doi: 10.3389/fcvm.2019.00132. eCollection 2019. Front Cardiovasc Med. 2019. PMID: 31572732 Free PMC article. Review.
-
Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein.J Cell Mol Med. 2009 Feb;13(2):373-87. doi: 10.1111/j.1582-4934.2008.00495.x. Epub 2008 Sep 13. J Cell Mol Med. 2009. PMID: 18793351 Free PMC article.
-
Thrombin and vascular inflammation.Mol Cell Biochem. 2012 Jan;359(1-2):301-13. doi: 10.1007/s11010-011-1024-x. Epub 2011 Aug 23. Mol Cell Biochem. 2012. PMID: 21858738 Review.
-
Circulating blood cells and extracellular vesicles in acute cardioprotection.Cardiovasc Res. 2019 Jun 1;115(7):1156-1166. doi: 10.1093/cvr/cvy314. Cardiovasc Res. 2019. PMID: 30590395 Free PMC article. Review.
References
-
- Chong AJ, Pohlman TH, Hampton CR, Shimamoto A, Mackman N, Verrier ED. Tissue factor and thrombin mediate myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2003;75:S649–S655. - PubMed
-
- Covic L, Misra M, Badar J, Singh C, Kuliopulos A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat Med. 2002;8:1161–1165. - PubMed
-
- Deguchi H, Takeya H, Urano H, Gabazza EC, Zhou H, Suzuki K. Adenosine regulates tissue factor expression on endothelial cells. Thromb Res. 1998;91:57–64. - PubMed
-
- D’Hondt C, Srinivas SP, Vereecke J, Himpens B. Adenosine opposes thrombin-induced inhibition of intercellular calcium wave in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2007;48:1518–1527. - PubMed
-
- Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation. 1997;96:2656–2666. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
