

Kaposi's sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications
- PMID: 18057235
- PMCID: PMC2258737
- DOI: 10.1128/JVI.00873-07
Kaposi's sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications
Abstract
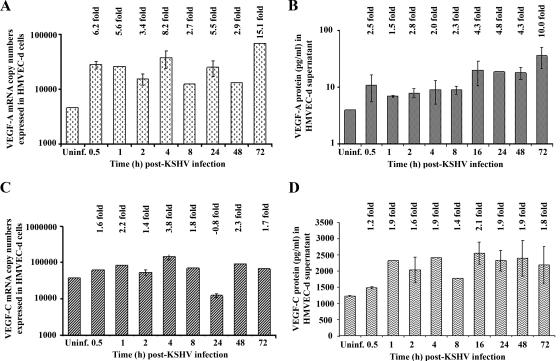
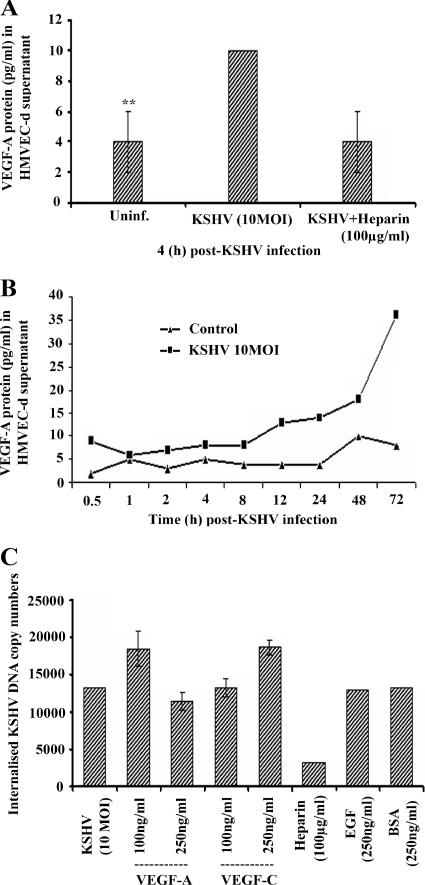
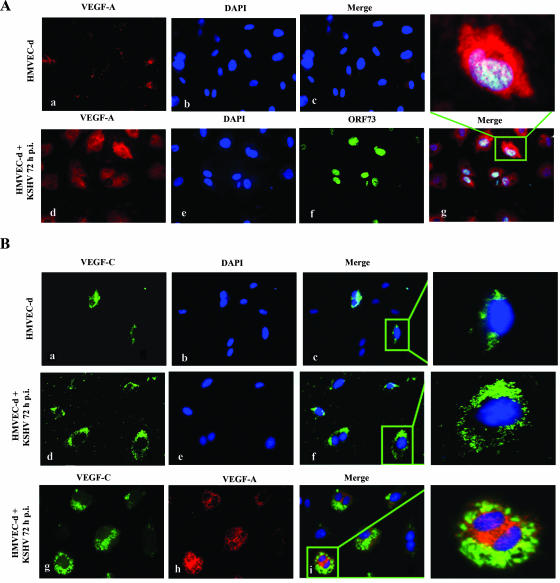
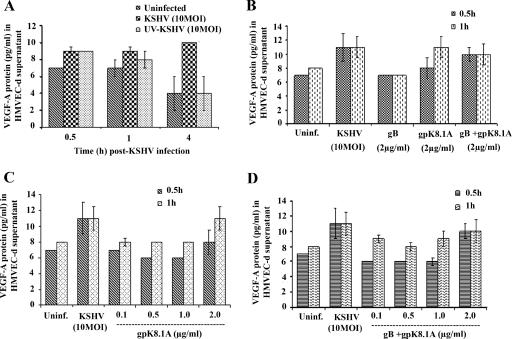
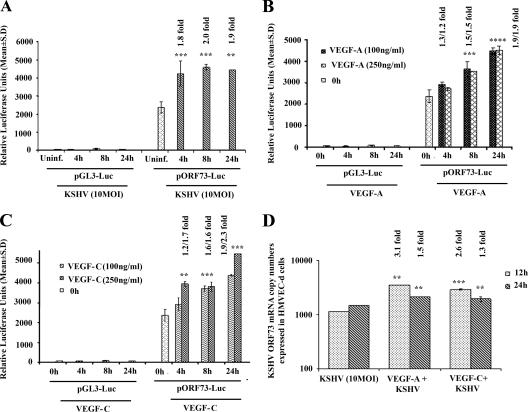
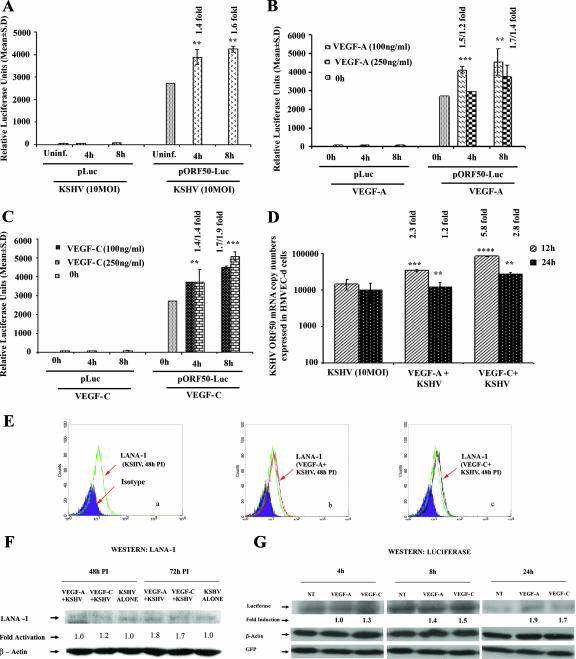
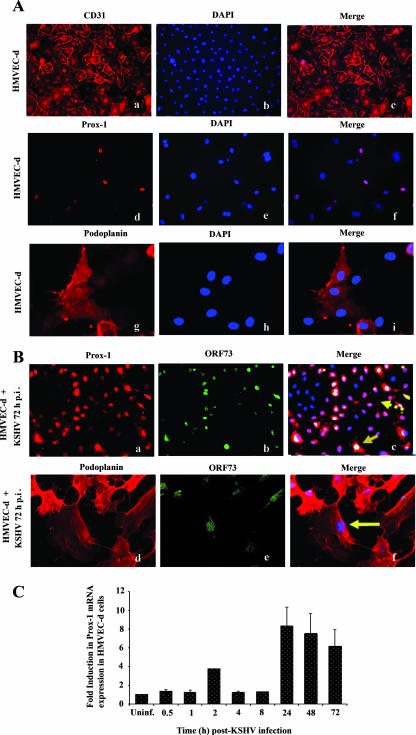
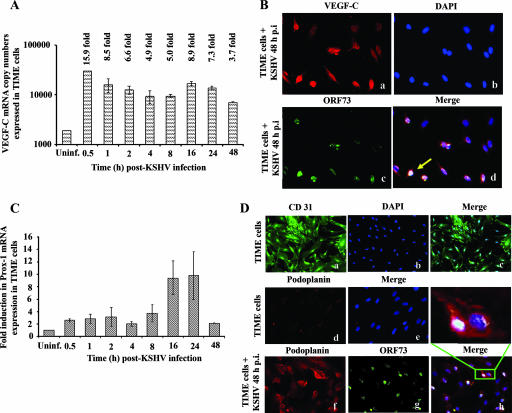
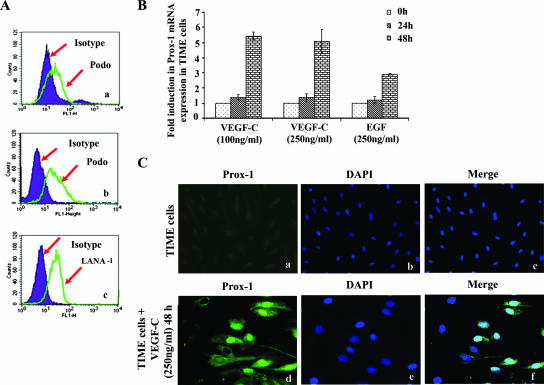
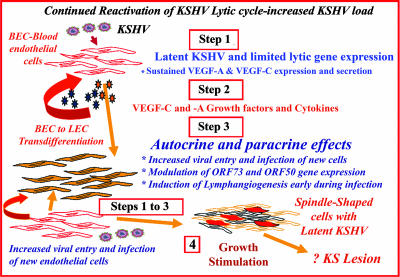
Kaposi's sarcoma (KS), a vascular tumor associated with human immunodeficiency virus type 1 infection, is characterized by spindle-shaped endothelial cells, inflammatory cells, cytokines, growth and angiogenic factors, and angiogenesis. KS spindle cells are believed to be of the lymphatic endothelial cell (LEC) type. Kaposi's sarcoma-associated herpesvirus (KSHV, or human herpesvirus 8) is etiologically linked to KS, and in vitro KSHV infection of primary human dermal microvascular endothelial cells (HMVEC-d) is characterized by the induction of preexisting host signal cascades, sustained expression of latency-associated genes, transient expression of a limited number of lytic genes, sustained induction of NF-kappaB and several cytokines, and growth and angiogenic factors. KSHV induced robust vascular endothelial growth factor A (VEGF-A) and VEGF-C gene expression as early as 30 min postinfection (p.i.) in serum-starved HMVEC-d, which was sustained throughout the observation period of 72 h p.i. Significant amounts of VEGF-A and -C were also detected in the culture supernatant of infected cells. VEGF-A and -C were also induced by UV-inactivated KSHV and envelope glycoprotein gpK8.1A, thus suggesting a role for virus entry stages in the early induction of VEGF and requirement of KSHV viral gene expression for sustained induction. Exogenous addition of VEGF-A and -C increased KSHV DNA entry into target cells and moderately increased latent ORF73 and lytic ORF50 promoter activation and gene expression. KSHV infection also induced the expression of lymphatic markers Prox-1 and podoplanin as early as 8 h p.i., and a paracrine effect was seen in the neighboring uninfected cells. Similar observations were also made in the pure blood endothelial cell (BEC)-TIME cells, thus suggesting that commitment to the LEC phenotype is induced early during KSHV infection of blood endothelial cells. Treatment with VEGF-C alone also induced Prox-1 expression in the BEC-TIME cells. Collectively, these studies show that the in vitro microenvironments of KSHV-infected endothelial cells are enriched, with VEGF-A and -C molecules playing key roles in KSHV biology, such as increased infection and gene expression, as well as in angiogenesis and lymphangiogenesis, thus recapitulating the microenvironment of early KS lesions.
Figures
Similar articles
-
ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection.J Virol. 2005 Aug;79(16):10308-29. doi: 10.1128/JVI.79.16.10308-10329.2005. J Virol. 2005. PMID: 16051824 Free PMC article.
-
Cyclooxygenase 2 induced by Kaposi's sarcoma-associated herpesvirus early during in vitro infection of target cells plays a role in the maintenance of latent viral gene expression.J Virol. 2006 Jul;80(13):6534-52. doi: 10.1128/JVI.00231-06. J Virol. 2006. PMID: 16775340 Free PMC article.
-
Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression.J Virol. 2007 Apr;81(8):3949-68. doi: 10.1128/JVI.02333-06. Epub 2007 Feb 7. J Virol. 2007. PMID: 17287275 Free PMC article.
-
Kaposi's sarcoma herpesvirus-induced endothelial cell reprogramming supports viral persistence and contributes to Kaposi's sarcoma tumorigenesis.Curr Opin Virol. 2017 Oct;26:156-162. doi: 10.1016/j.coviro.2017.09.002. Epub 2017 Oct 12. Curr Opin Virol. 2017. PMID: 29031103 Review.
-
KSHV Entry and Trafficking in Target Cells-Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics.Viruses. 2016 Nov 14;8(11):305. doi: 10.3390/v8110305. Viruses. 2016. PMID: 27854239 Free PMC article. Review.
Cited by
-
KSHV-Mediated Angiogenesis in Tumor Progression.Viruses. 2016 Jul 20;8(7):198. doi: 10.3390/v8070198. Viruses. 2016. PMID: 27447661 Free PMC article. Review.
-
Latent KSHV infection increases the vascular permeability of human endothelial cells.Blood. 2011 Nov 10;118(19):5344-54. doi: 10.1182/blood-2011-03-341552. Epub 2011 Aug 31. Blood. 2011. PMID: 21881052 Free PMC article.
-
Kaposi's sarcoma-associated herpesvirus glycoproteins B and K8.1 regulate virion egress and synthesis of vascular endothelial growth factor and viral interleukin-6 in BCBL-1 cells.J Virol. 2010 Feb;84(4):1704-14. doi: 10.1128/JVI.01889-09. Epub 2009 Dec 2. J Virol. 2010. PMID: 19955303 Free PMC article.
-
Inflammatory stress and sarcomagenesis: a vicious interplay.Cell Stress Chaperones. 2014 Jan;19(1):1-13. doi: 10.1007/s12192-013-0449-4. Epub 2013 Aug 27. Cell Stress Chaperones. 2014. PMID: 24046208 Free PMC article. Review.
-
DLX1008 (brolucizumab), a single-chain anti-VEGF-A antibody fragment with low picomolar affinity, leads to tumor involution in an in vivo model of Kaposi Sarcoma.PLoS One. 2020 May 14;15(5):e0233116. doi: 10.1371/journal.pone.0233116. eCollection 2020. PLoS One. 2020. PMID: 32407363 Free PMC article.
References
-
- Akula, S. M., N. P. Pramod, F.-Z. Wang, and B. Chandran. 2002. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108407-419. - PubMed
-
- Akula, S. M., F.-Z. Wang, J. Vieira, and B. Chandran. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282245-255. - PubMed
-
- Albini, A., I. Paglieri, G. Orengo, S. Carlone, M. G. Aluigi, R. DeMarchi, C. Matteucci, A. Mantovani, F. Carozzi, S. Donini, and R. Benelli. 1997. The beta-core fragment of human chorionic gonadotrophin inhibits growth of Kaposi's sarcoma-derived cells and a new immortalized Kaposi's sarcoma cell line. AIDS 11713-721. - PubMed
-
- Alitalo, K., T. Tammela, and T. V. Petrova. 2005. Lymphangiogenesis in development and human disease. Nature 438946-953. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
