

A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response
- PMID: 18060054
- PMCID: PMC2082660
- DOI: 10.1371/journal.pone.0001258
A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response
Abstract
Background: SCN5A encodes the alpha-subunit (Na(v)1.5) of the principle Na(+) channel in the human heart. Genetic lesions in SCN5A can cause congenital long QT syndrome (LQTS) variant 3 (LQT-3) in adults by disrupting inactivation of the Na(v)1.5 channel. Pharmacological targeting of mutation-altered Na(+) channels has proven promising in developing a gene-specific therapeutic strategy to manage specifically this LQTS variant. SCN5A mutations that cause similar channel dysfunction may also contribute to sudden infant death syndrome (SIDS) and other arrhythmias in newborns, but the prevalence, impact, and therapeutic management of SCN5A mutations may be distinct in infants compared with adults.
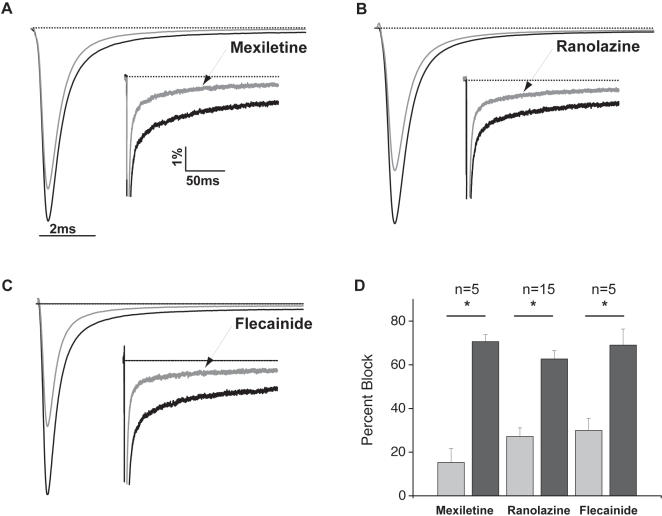
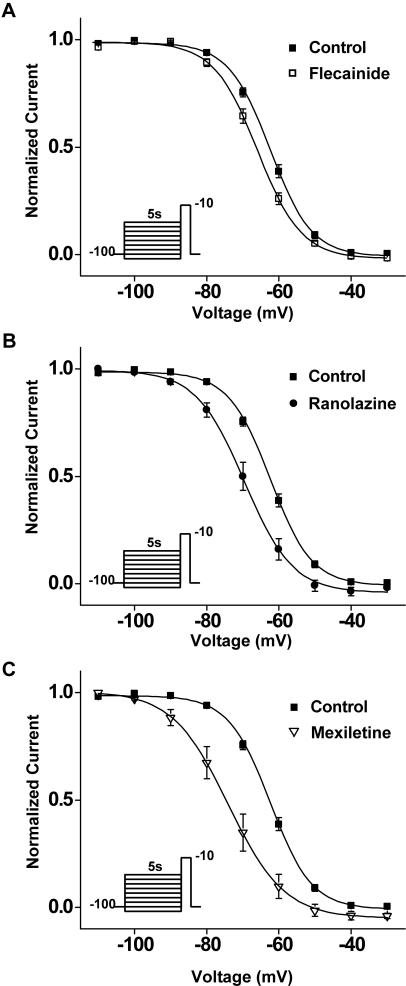
Methods and results: Here, in a multidisciplinary approach, we report a de novo SCN5A mutation (F1473C) discovered in a newborn presenting with extreme QT prolongation and differential responses to the Na(+) channel blockers flecainide and mexiletine. Our goal was to determine the Na(+) channel phenotype caused by this severe mutation and to determine whether distinct effects of different Na(+) channel blockers on mutant channel activity provide a mechanistic understanding of the distinct therapeutic responsiveness of the mutation carrier. Sequence analysis of the proband revealed the novel missense SCN5A mutation (F1473C) and a common variant in KCNH2 (K897T). Patch clamp analysis of HEK 293 cells transiently transfected with wild-type or mutant Na(+) channels revealed significant changes in channel biophysics, all contributing to the proband's phenotype as predicted by in silico modeling. Furthermore, subtle differences in drug action were detected in correcting mutant channel activity that, together with both the known genetic background and age of the patient, contribute to the distinct therapeutic responses observed clinically.
Significance: The results of our study provide further evidence of the grave vulnerability of newborns to Na(+) channel defects and suggest that both genetic background and age are particularly important in developing a mutation-specific therapeutic personalized approach to manage disorders in the young.
Conflict of interest statement
Figures






References
-
- Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, et al. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. - PubMed
-
- Kass RS, Moss AJ. Mutation-specific pharmacology of the long QT syndrome. Handb Exp Pharmacol. 2006:287–304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
