

Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome
- PMID: 18067674
- PMCID: PMC2234399
- DOI: 10.1186/1750-1172-2-48
Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome
Abstract
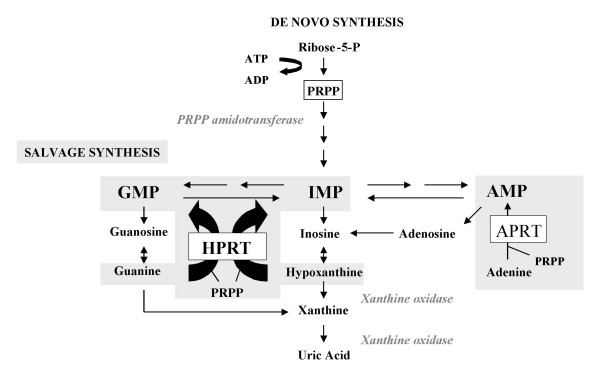
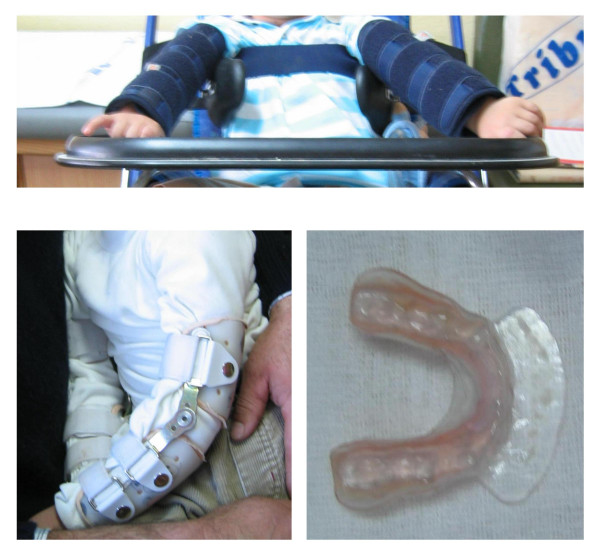
Deficiency of hypoxanthine-guanine phosphoribosyltransferase (HPRT) activity is an inborn error of purine metabolism associated with uric acid overproduction and a continuum spectrum of neurological manifestations depending on the degree of the enzymatic deficiency. The prevalence is estimated at 1/380,000 live births in Canada, and 1/235,000 live births in Spain. Uric acid overproduction is present inall HPRT-deficient patients and is associated with lithiasis and gout. Neurological manifestations include severe action dystonia, choreoathetosis, ballismus, cognitive and attention deficit, and self-injurious behaviour. The most severe forms are known as Lesch-Nyhan syndrome (patients are normal at birth and diagnosis can be accomplished when psychomotor delay becomes apparent). Partial HPRT-deficient patients present these symptoms with a different intensity, and in the least severe forms symptoms may be unapparent. Megaloblastic anaemia is also associated with the disease. Inheritance of HPRT deficiency is X-linked recessive, thus males are generally affected and heterozygous female are carriers (usually asymptomatic). Human HPRT is encoded by a single structural gene on the long arm of the X chromosome at Xq26. To date, more than 300 disease-associated mutations in the HPRT1 gene have been identified. The diagnosis is based on clinical and biochemical findings (hyperuricemia and hyperuricosuria associated with psychomotor delay), and enzymatic (HPRT activity determination in haemolysate, intact erythrocytes or fibroblasts) and molecular tests. Molecular diagnosis allows faster and more accurate carrier and prenatal diagnosis. Prenatal diagnosis can be performed with amniotic cells obtained by amniocentesis at about 15-18 weeks' gestation, or chorionic villus cells obtained at about 10-12 weeks' gestation. Uric acid overproduction can be managed by allopurinol treatment. Doses must be carefully adjusted to avoid xanthine lithiasis. The lack of precise understanding of the neurological dysfunction has precluded development of useful therapies. Spasticity, when present, and dystonia can be managed with benzodiazepines and gamma-aminobutyric acid inhibitors such as baclofen. Physical rehabilitation, including management of dysarthria and dysphagia, special devices to enable hand control, appropriate walking aids, and a programme of posture management to prevent deformities are recommended. Self-injurious behaviour must be managed by a combination of physical restraints, behavioural and pharmaceutical treatments.
Figures
References
-
- Kelley WN, Greene ML, Rosenbloom FM, Henderson JF, Seegmiller JE. Hypoxanthine-guanine phosphoribosyltransferase deficiency in gout. Ann Intern Med. 1969;70:155–206. - PubMed
-
- Catel W, Schmidt J. On familial gouty diathesis associated with cerebral and renal symptoms in a small child. Dtsch Med Wochenschr. 1959;84:2145–2147. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
