

Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila
- PMID: 18073425
- PMCID: PMC2219485
- DOI: 10.1534/genetics.107.080226
Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila
Abstract
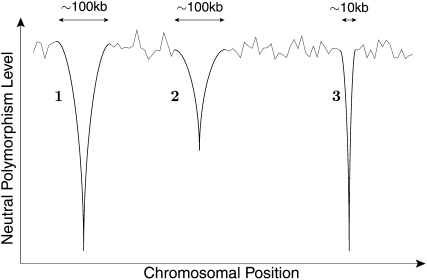
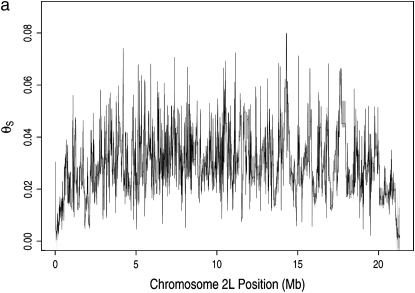
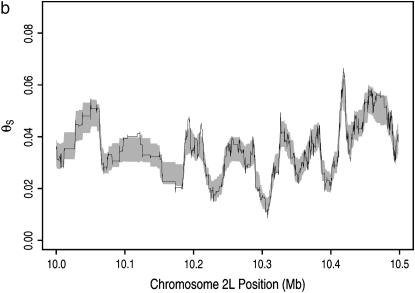
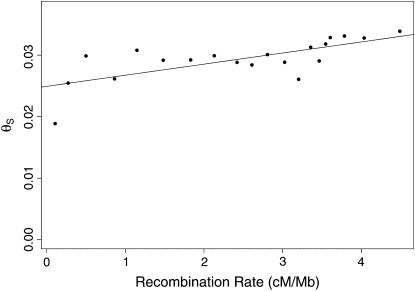
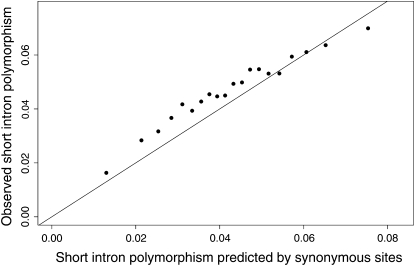
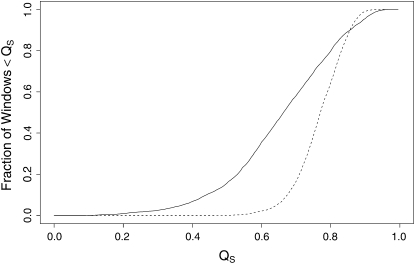
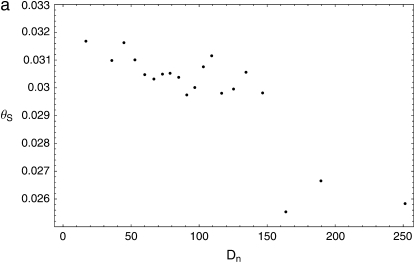
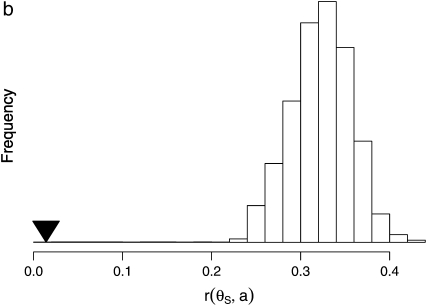
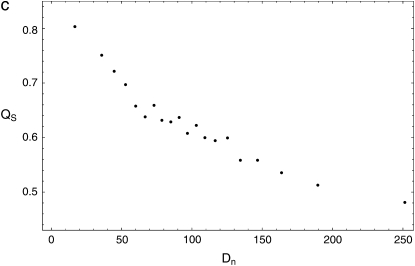
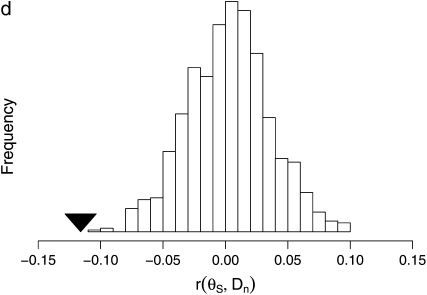
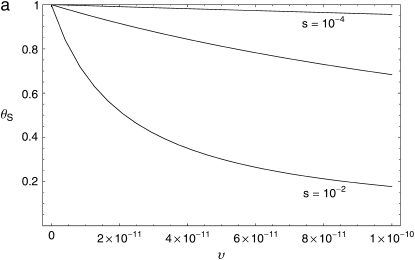
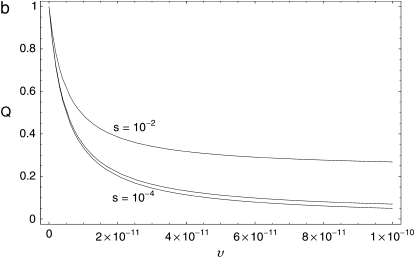
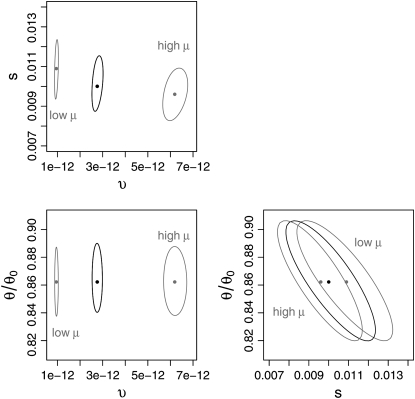
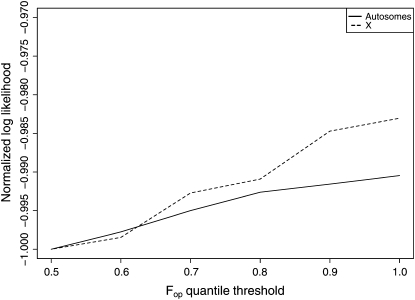
The effect of recurrent selective sweeps is a spatially heterogeneous reduction in neutral polymorphism throughout the genome. The pattern of reduction depends on the selective advantage and recurrence rate of the sweeps. Because many adaptive substitutions responsible for these sweeps also contribute to nonsynonymous divergence, the spatial distribution of nonsynonymous divergence also reflects the distribution of adaptive substitutions. Thus, the spatial correspondence between neutral polymorphism and nonsynonymous divergence may be especially informative about the process of adaptation. Here we study this correspondence using genomewide polymorphism data from Drosophila simulans and the divergence between D. simulans and D. melanogaster. Focusing on highly recombining portions of the autosomes, at a spatial scale appropriate to the study of selective sweeps, we find that neutral polymorphism is both lower and, as measured by a new statistic Q(S), less homogeneous where nonsynonymous divergence is higher and that the spatial structure of this correlation is best explained by the action of strong recurrent selective sweeps. We introduce a method to infer, from the spatial correspondence between polymorphism and divergence, the rate and selective strength of adaptation. Our results independently confirm a high rate of adaptive substitution (approximately 1/3000 generations) and newly suggest that many adaptations are of surprisingly great selective effect (approximately 1%), reducing the effective population size by approximately 15% even in highly recombining regions of the genome.
Figures
References
-
- Akashi, H., and A. Eyre-Walker, 1998. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 8 688–693. - PubMed
-
- Aminetzach, Y. T., J. M. Macpherson and D. A. Petrov, 2005. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science 309 764–767. - PubMed
-
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437 1149–1152. - PubMed
-
- Bates, D. M., and D. G. Watts, 1988. Nonlinear Regression Analysis and Its Applications. Wiley, New York.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
