

Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA
- PMID: 18079180
- PMCID: PMC2113034
- DOI: 10.1101/gad.455407
Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA
Abstract
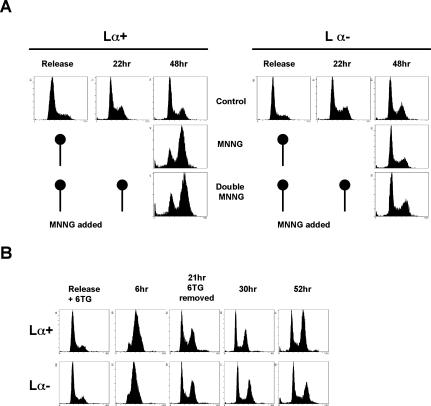
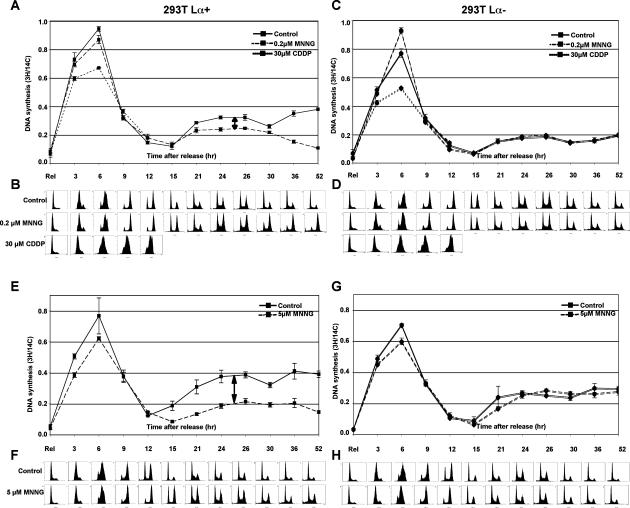
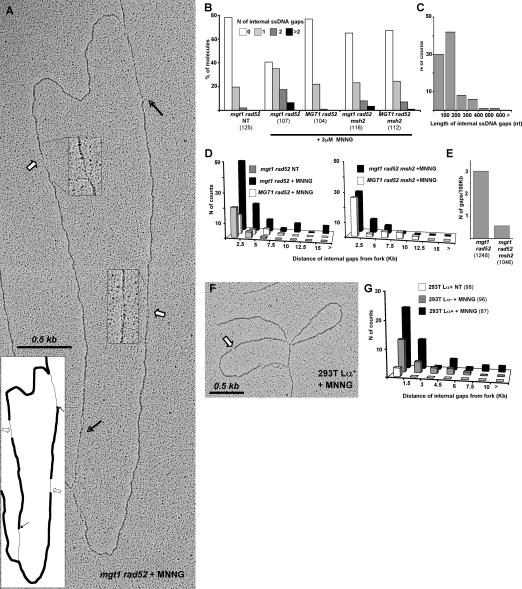
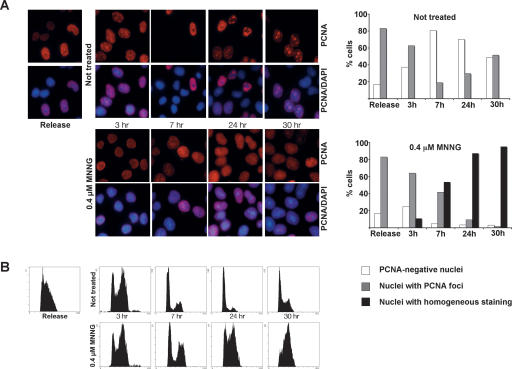
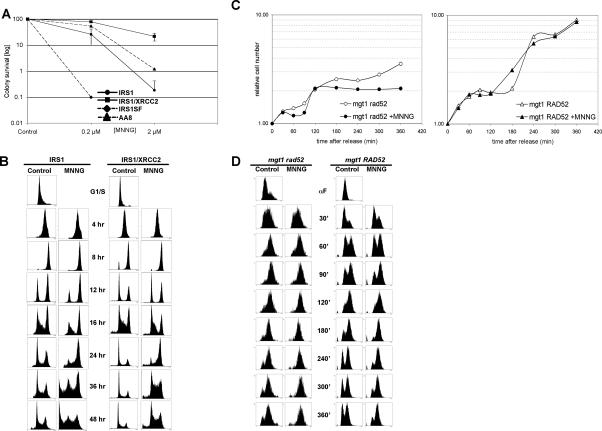
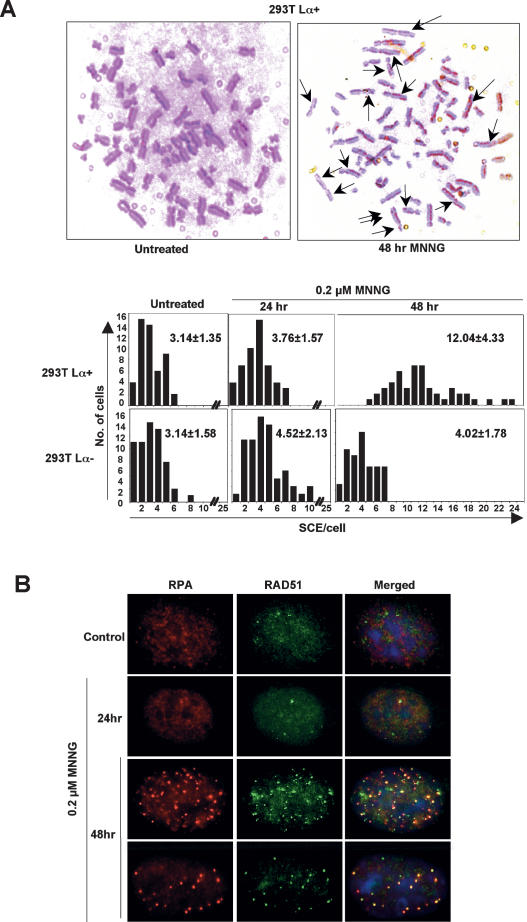
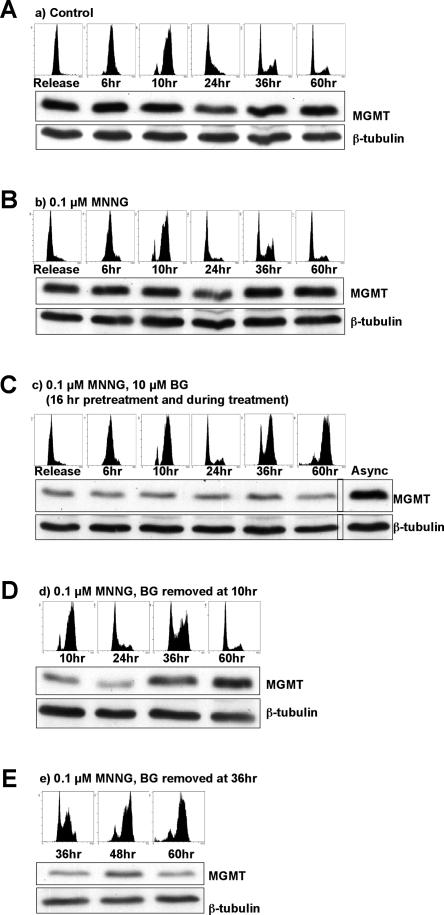
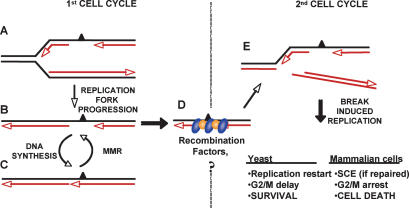
O(6)-Methylguanine ((Me)G) is a highly cytotoxic DNA modification generated by S(N)1-type methylating agents. Despite numerous studies implicating DNA replication, mismatch repair (MMR), and homologous recombination (HR) in (Me)G toxicity, its mode of action has remained elusive. We studied the molecular transactions in the DNA of yeast and mammalian cells treated with N-methyl-N'-nitro-N-nitrosoguanidine (MNNG). Although replication fork progression was unaffected in the first cell cycle after treatment, electron microscopic analysis revealed an accumulation of (Me)G- and MMR-dependent single-stranded DNA (ssDNA) gaps in newly replicated DNA. Progression into the second cell cycle required HR, while the following G(2) arrest required the continued presence of (Me)G. Yeast cells overcame this block, while mammalian cells generally failed to recover, and those that did contained multiple sister chromatid exchanges. Notably, the arrest could be abolished by removal of (Me)G after the first S phase. These new data provide compelling support for the hypothesis that MMR attempts to correct (Me)G/C or (Me)G/T mispairs arising during replication. Due to the persistence of (Me)G in the exposed template strand, repair synthesis cannot take place, which leaves single-stranded gaps behind the replication fork. During the subsequent S phase, these gaps cause replication fork collapse and elicit recombination and cell cycle arrest.
Figures
References
-
- Berardini M., Mazurek A., Fishel R., Mazurek A., Fishel R., Fishel R. The effect of O6-methylguanine DNA adducts on the adenosine nucleotide switch functions of hMSH2–hMSH6 and hMSH2–hMSH3. J. Biol. Chem. 2000;275:27851–27857. - PubMed
-
- Cejka P., Stojic L., Mojas N., Russell A.M., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J., Stojic L., Mojas N., Russell A.M., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J., Mojas N., Russell A.M., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J., Russell A.M., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J., Heinimann K., Cannavo E., di Pietro M., Marra G., Jiricny J., Cannavo E., di Pietro M., Marra G., Jiricny J., di Pietro M., Marra G., Jiricny J., Marra G., Jiricny J., Jiricny J. Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J. 2003;22:2245–2254. - PMC - PubMed
-
- Cejka P., Mojas N., Gillet L., Schar P., Jiricny J., Mojas N., Gillet L., Schar P., Jiricny J., Gillet L., Schar P., Jiricny J., Schar P., Jiricny J., Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr. Biol. 2005;15:1395–1400. - PubMed
-
- Clark A.B., Valle F., Drotschmann K., Gary R.K., Kunkel T.A., Valle F., Drotschmann K., Gary R.K., Kunkel T.A., Drotschmann K., Gary R.K., Kunkel T.A., Gary R.K., Kunkel T.A., Kunkel T.A. Functional interaction of proliferating cell nuclear antigen with MSH2–MSH6 and MSH2–MSH3 complexes. J. Biol. Chem. 2000;275:36498–36501. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials