

The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice
- PMID: 18079970
- PMCID: PMC2129240
- DOI: 10.1172/JCI33255
The MEF2D transcription factor mediates stress-dependent cardiac remodeling in mice
Abstract
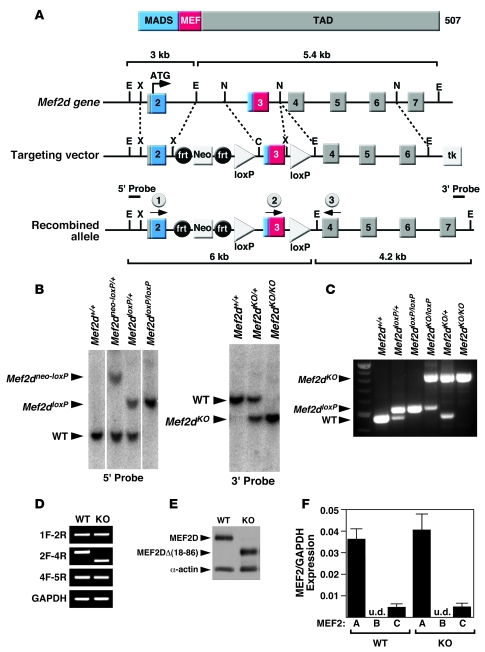
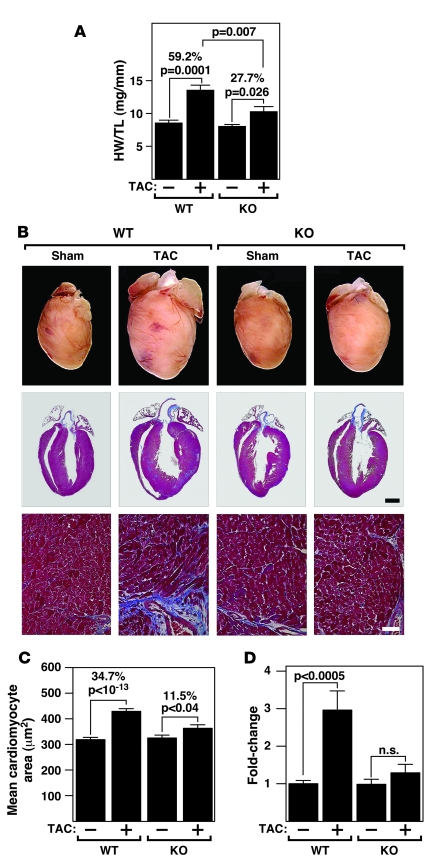
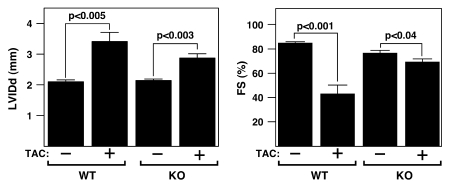
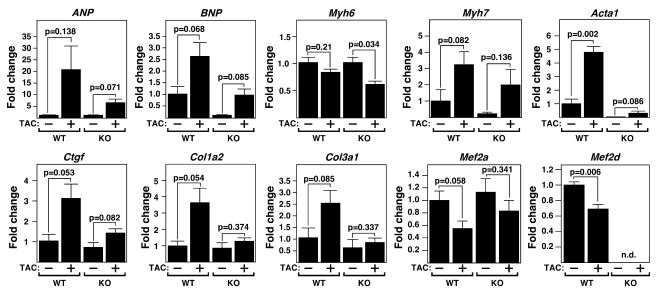
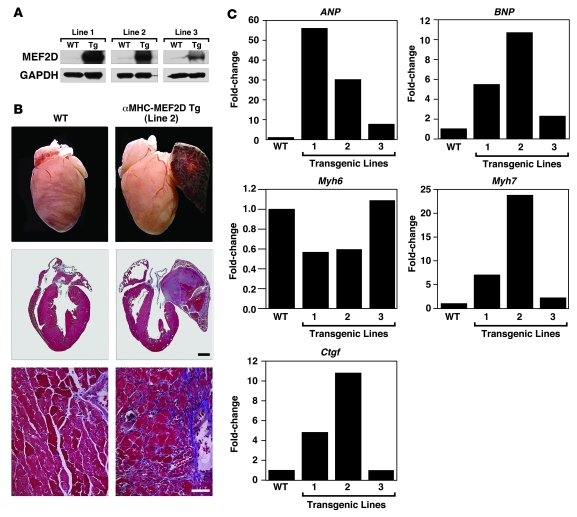
The adult heart responds to excessive neurohumoral signaling and workload by a pathological growth response characterized by hypertrophy of cardiomyocytes and activation of a fetal program of cardiac gene expression. These responses culminate in diminished pump function, ventricular dilatation, wall thinning, and fibrosis, and can result in sudden death. Myocyte enhancer factor-2 (MEF2) transcription factors serve as targets of the signaling pathways that drive pathological cardiac remodeling, but the requirement for MEF2 factors in the progression of heart disease in vivo has not been determined. MEF2A and MEF2D are the primary MEF2 factors expressed in the adult heart. To specifically determine the role of MEF2D in pathological cardiac remodeling, we generated mice with a conditional MEF2D allele. MEF2D-null mice were viable, but were resistant to cardiac hypertrophy, fetal gene activation, and fibrosis in response to pressure overload and beta-chronic adrenergic stimulation. Furthermore, we show in a transgenic mouse model that forced overexpression of MEF2D was sufficient to drive the fetal gene program and pathological remodeling of the heart. These results reveal a unique and important function for MEF2D in stress-dependent cardiac growth and reprogramming of gene expression in the adult heart.
Figures

References
-
- Czubryt M.P., Olson E.N. Balancing contractility and energy production: the role of myocyte enhancer factor 2 (MEF2) in cardiac hypertrophy. Recent Prog. Horm. Res. 2004;59:105–124. - PubMed
-
- Frey N., Katus H.A., Olson E.N., Hill J.A. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. - PubMed
-
- Frey N., Olson E.N. Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 2003;65:45–79. - PubMed
-
- Backs J., Olson E.N. Control of cardiac growth by histone acetylation/deacetylation. Circ. Res. 2006;98:15–24. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
