

Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome
- PMID: 18087273
- PMCID: PMC2361463
- DOI: 10.1038/sj.bjc.6604156
Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome
Abstract
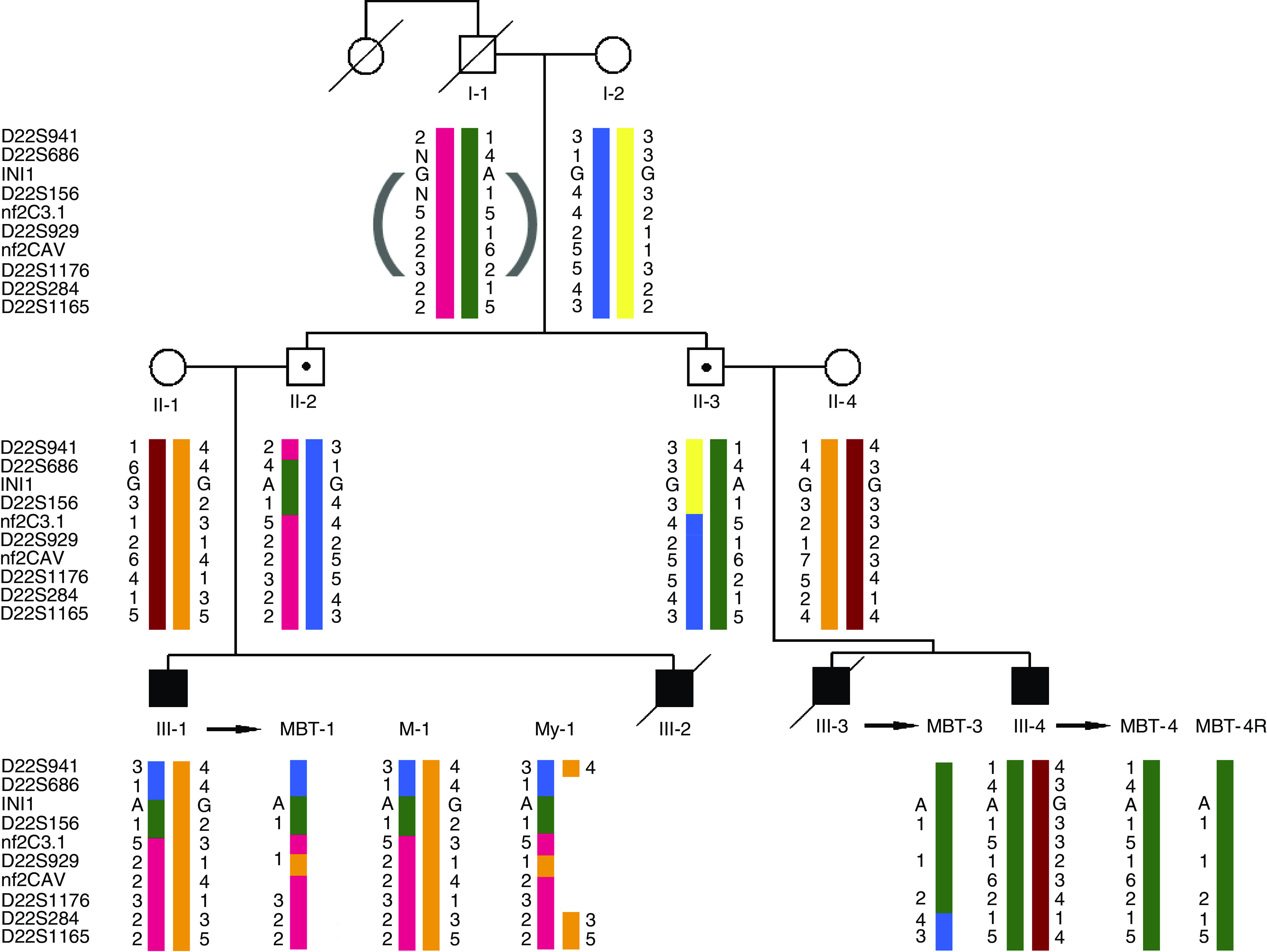
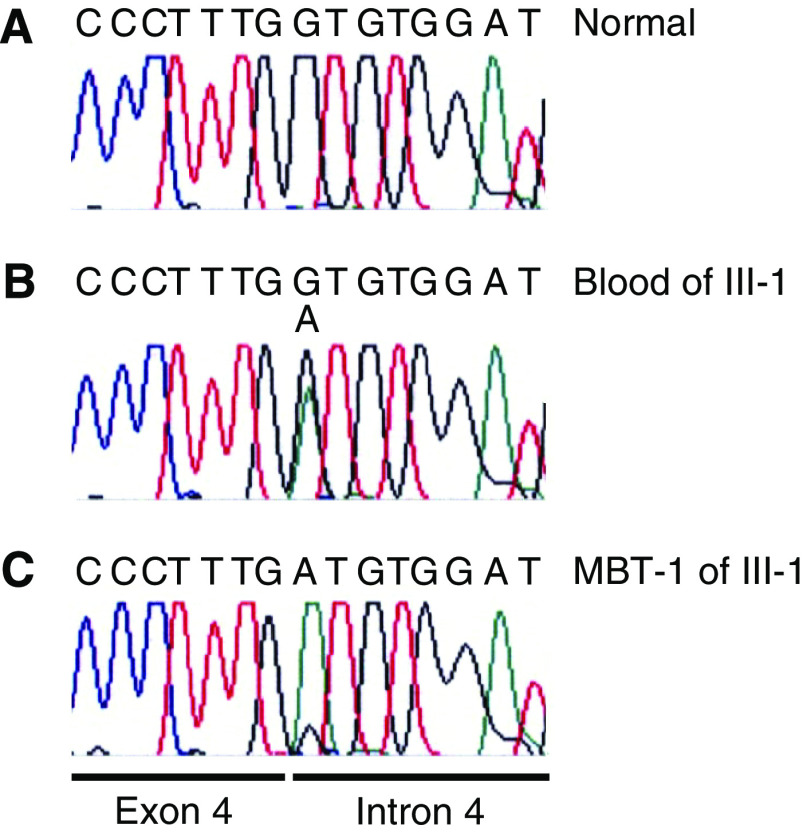
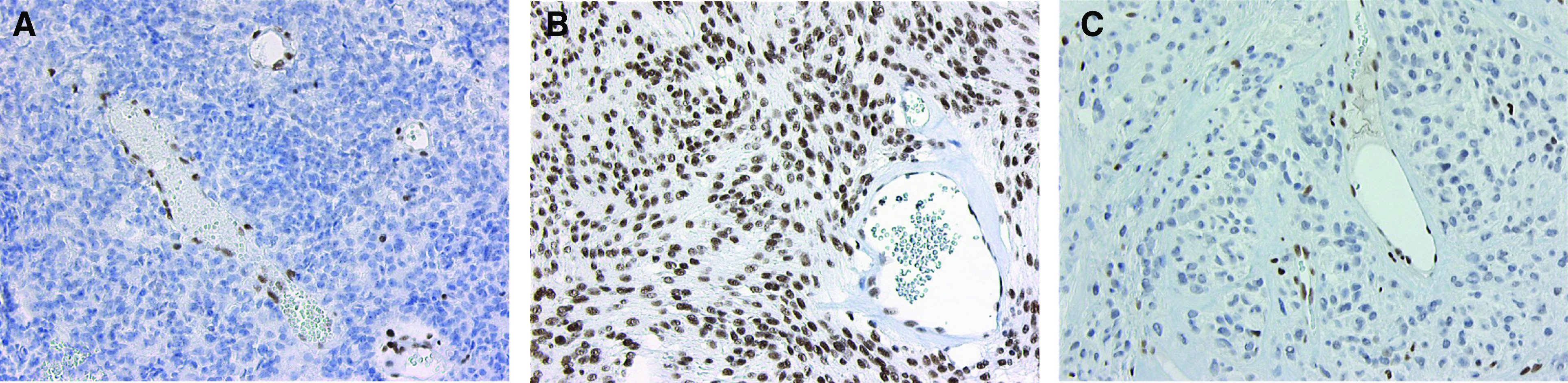
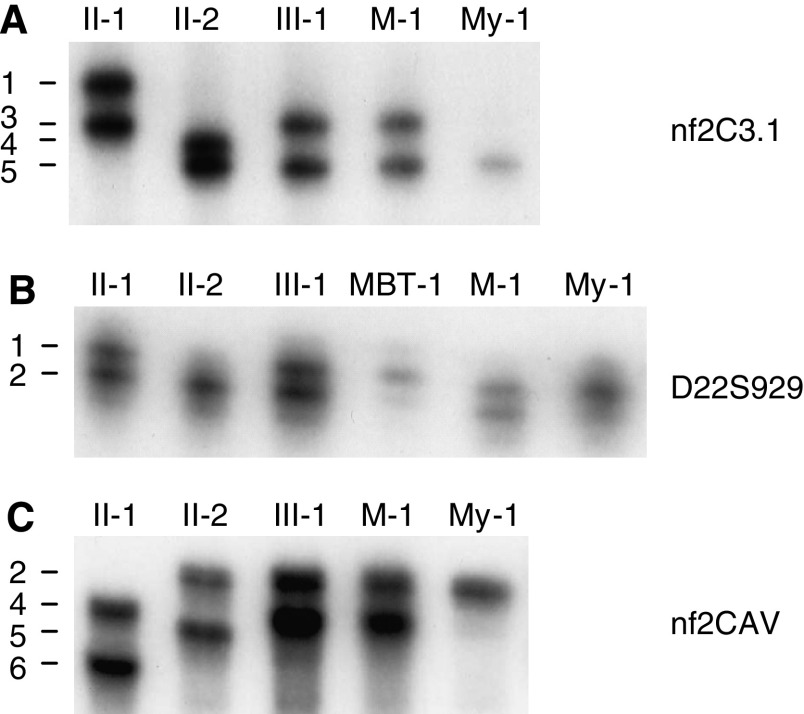
Rhabdoid tumour predisposition syndrome (RTPS) is a rare syndrome caused by inheritance of a mutated INI1 gene for which only two multigeneration families have been reported. To further characterise the genotype and phenotype of RTPS, we present a third family in which at least three cousins developed an atypical teratoid/rhabdoid tumour (AT/RT) at a young age. Two of these patients showed unusual long survival, and one of these developed an intracranial meningioma and a myoepithelioma of the lip in adulthood. Mutation analysis of INI1 revealed a germline G>A mutation in the donor splice site of exon 4 (c.500+1G>A) in the patients and in their unaffected fathers. This mutation prevents normal splicing and concomitantly generates a stop codon, resulting in nonsense-mediated mRNA decay. Biallelic inactivation of INI1 in the tumours, except for the meningioma, was confirmed by absence of nuclear INI1-protein staining. The myoepithelioma of one of the patients carried an identical somatic rearrangement in the NF2 gene as the AT/RT, indicating that both tumours originated from a common precursor cell. In conclusion, this study demonstrates for the first time transmission of a germline INI1-mutation in a RTPS family via nonpenetrant males, long-term survival of two members of this family with an AT/RT, and involvement of INI1 in the pathogenesis of myoepithelioma.
Figures
References
-
- Biegel JA (2006) Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 20: 1–7 - PubMed
-
- Bijlsma EK, Voesten AM, Bijleveld EH, Troost D, Westerveld A, Merel P, Thomas G, Hulsebos TJ (1995) Molecular analysis of genetic changes in ependymomas. Genes Chromosomes Cancer 13: 272–277 - PubMed
-
- Dimopoulos VG, Fountas KN, Robinson JS (2006) Familial intracranial ependymomas. Report of three cases in a family and review of the literature. Neurosurg Focus 20: 1–5 - PubMed
-
- Favre M, Butticaz C, Stevenson B, Jongeneel CV, Telenti A (2003) High frequency of alternative splicing of human genes participating in the HIV-1 life cycle: a model using TSG101, betaTrCP, PPIA, INI1, NAF1, and PML. J Acquir Immune Defic Syndr 34: 127–133 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
