

DNA damage-induced cell death: lessons from the central nervous system
- PMID: 18087290
- PMCID: PMC2626635
- DOI: 10.1038/cr.2007.110
DNA damage-induced cell death: lessons from the central nervous system
Abstract
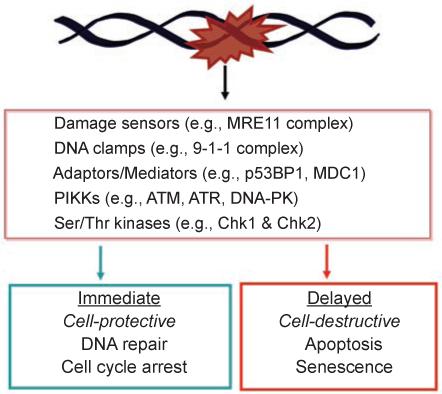
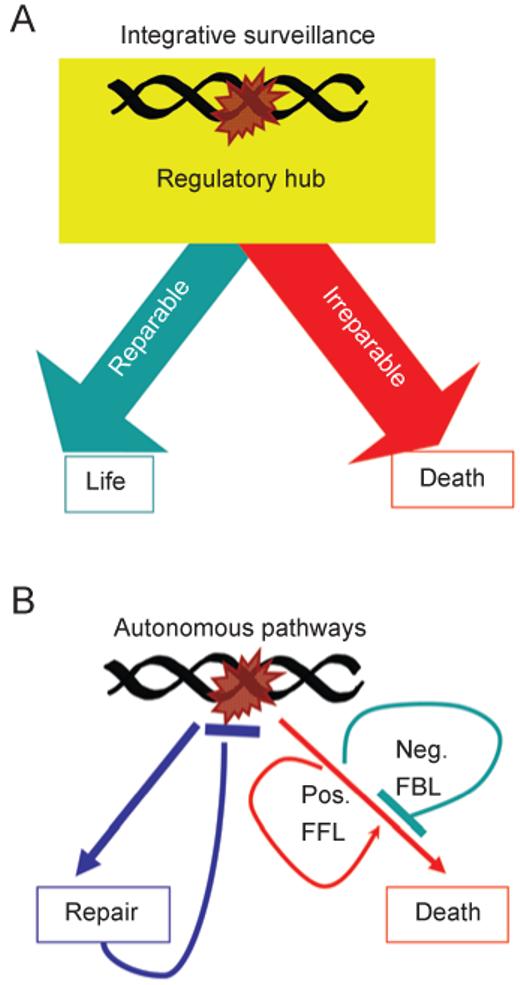
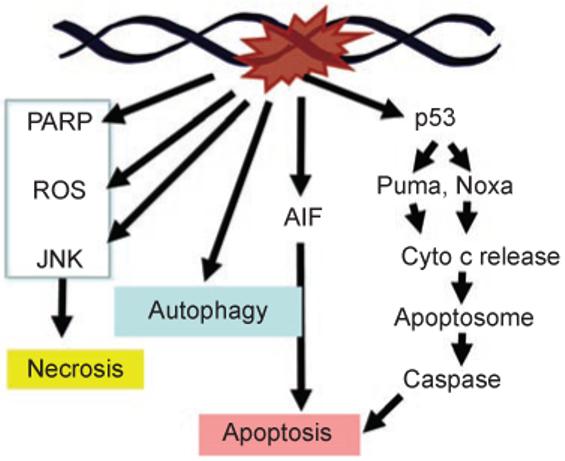
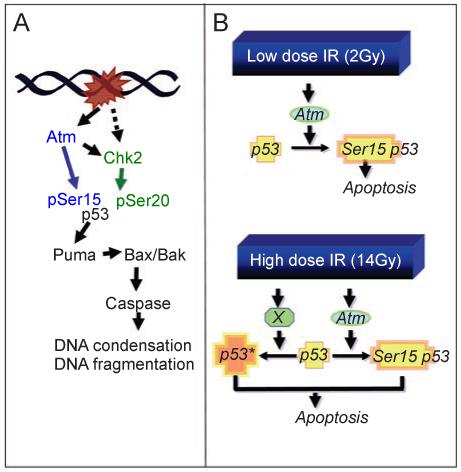
DNA damage can, but does not always, induce cell death. While several pathways linking DNA damage signals to mitochondria-dependent and -independent death machineries have been elucidated, the connectivity of these pathways is subject to regulation by multiple other factors that are not well understood. We have proposed two conceptual models to explain the delayed and variable cell death response to DNA damage: integrative surveillance versus autonomous pathways. In this review, we discuss how these two models may explain the in vivo regulation of cell death induced by ionizing radiation (IR) in the developing central nervous system, where the death response is regulated by radiation dose, cell cycle status and neuronal development.
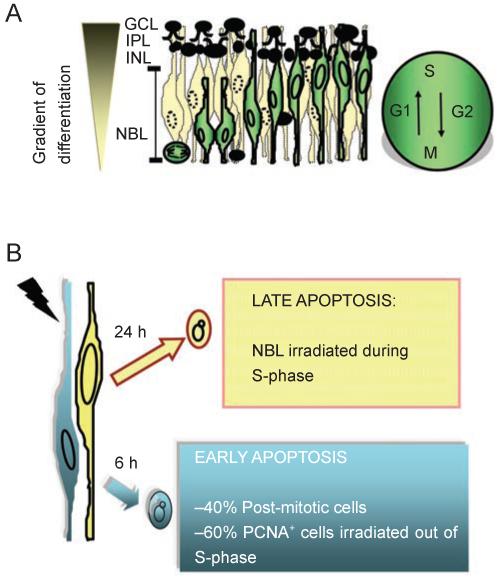
Figures
References
-
- Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–783. - PubMed
-
- Blank M, Shiloh Y. Programs for cell death: apoptosis is only one way to go. Cell Cycle. 2007;6:686–695. - PubMed
-
- Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. - PubMed
-
- Bentle MS, Bey EA, Dong Y, Reinicke KE, Boothman DA. New tricks for old drugs: the anticarcinogenic potential of DNA repair inhibitors. J Mol Histol. 2006;37:203–218. - PubMed
-
- Michod D, Widmann C. DNA-damage sensitizers: potential new therapeutical tools to improve chemotherapy. Crit Rev Oncol Hematol. 2007;63:160–171. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
