

doi: 10.1186/gb-2007-8-12-r269.
CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction
Affiliations
- PMID: 18096039
- PMCID: PMC2246271
- DOI: 10.1186/gb-2007-8-12-r269
Item in Clipboard
CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction
Genome Biol.
2007.
Abstract
We describe CONTRAST, a gene predictor which directly incorporates information from multiple alignments rather than employing phylogenetic models. This is accomplished through the use of discriminative machine learning techniques, including a novel training algorithm. We use a two-stage approach, in which a set of binary classifiers designed to recognize coding region boundaries is combined with a global model of gene structure. CONTRAST predicts exact coding region structures for 65% more human genes than the previous state-of-the-art method, misses 46% fewer exons and displays comparable gains in specificity.
Figures
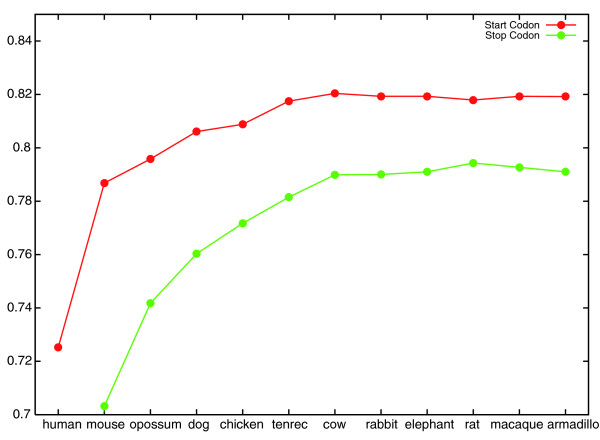
Start and stop codon classifier accuracy increases as informants are added. The graph shows the generalization accuracy of CONTRAST's start and stop codon classifiers as more informants are added. The x-axis labels indicate the most recently added informant. For example, at the point labeled 'chicken', the informant set consists of mouse, opossum, dog and chicken.
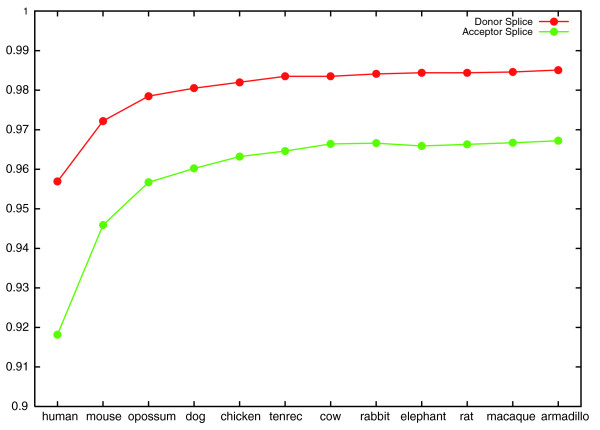
Splice site classifier accuracy increases as informants are added. The graph shows the generalization accuracy of CONTRAST's donor and acceptor splice site classifiers as more informants are added. The x-axis labels indicate the most recently added informant. For example, at the point labeled 'chicken', the informant set consists of mouse, opossum, dog and chicken.

Part of a typical set of input data. The input data consists of 13 rows. The first row contains sequence from the target genome, the second to twelfth rows contain aligned sequence from informant genomes and the last row encodes information about the alignments of ESTs to the target genome.
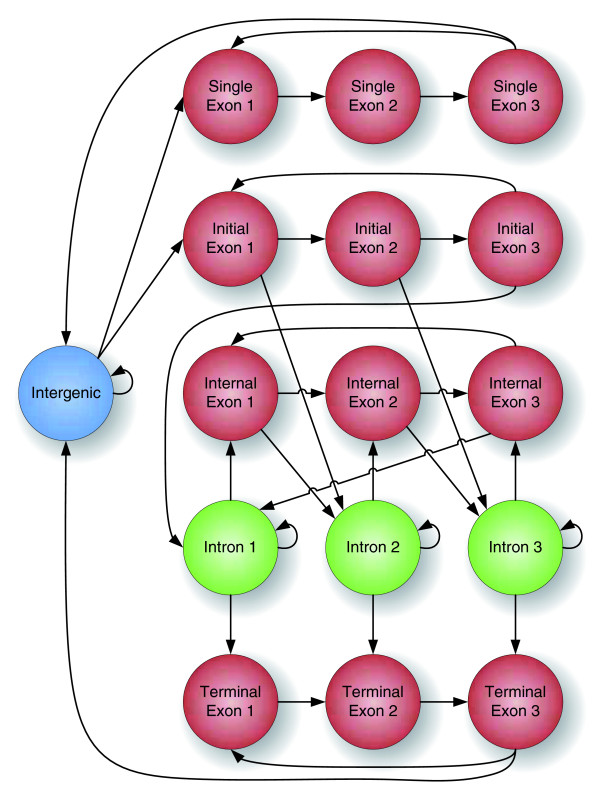
The structure of labelings in CONTRAST. Each node in the graph is a possible label for a single position in the target sequence. A labeling is legal if it corresponds to a path through the graph.
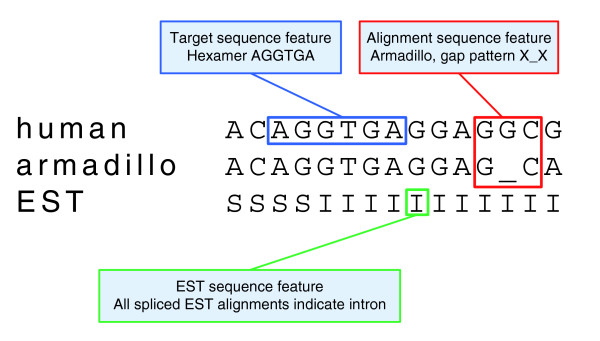
Features that score a label based on local sequence. CONTRAST contains three types of features for scoring a label based on local sequence: features based on hexamers in the target sequence (shown in blue), features based on a trimer in the target sequence and a trimer in an informant alignment (shown in red) and features based on a position in the EST sequence (shown in green).

Features that score coding region boundaries. CONTRAST contains two types of feature for scoring coding region boundaries. The first, shown in red, maps the output of a classifier to a score using a piecewise linear function learned during CRF training. In this example, the score from the GT splice donor classifier falls between the fourth and fifth control points for the function, with interpolation coefficients of 0.312 and 0.688. The second type of feature, shown in green, scores a coding region boundary based on the EST sequence characters that flank it.
Similar articles
-
Using ESTs to improve the accuracy of de novo gene prediction.BMC Bioinformatics. 2006 Jul 3;7:327. doi: 10.1186/1471-2105-7-327. BMC Bioinformatics. 2006. PMID: 16817966 Free PMC article.
-
Gene prediction: compare and CONTRAST.Genome Biol. 2007;8(12):233. doi: 10.1186/gb-2007-8-12-233. Genome Biol. 2007. PMID: 18096089 Free PMC article. Review.
-
Vertebrate gene finding from multiple-species alignments using a two-level strategy.Genome Biol. 2006;7 Suppl 1(Suppl 1):S6.1-12. doi: 10.1186/gb-2006-7-s1-s6. Epub 2006 Aug 7. Genome Biol. 2006. PMID: 16925840 Free PMC article.
-
SVM-Fold: a tool for discriminative multi-class protein fold and superfamily recognition.BMC Bioinformatics. 2007 May 22;8 Suppl 4(Suppl 4):S2. doi: 10.1186/1471-2105-8-S4-S2. BMC Bioinformatics. 2007. PMID: 17570145 Free PMC article.
-
GeneAlign: a coding exon prediction tool based on phylogenetical comparisons.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W280-4. doi: 10.1093/nar/gkl307. Nucleic Acids Res. 2006. PMID: 16845010 Free PMC article.
Cited by
-
Generalized centroid estimators in bioinformatics.PLoS One. 2011 Feb 18;6(2):e16450. doi: 10.1371/journal.pone.0016450. PLoS One. 2011. PMID: 21365017 Free PMC article.
-
mGene: accurate SVM-based gene finding with an application to nematode genomes.Genome Res. 2009 Nov;19(11):2133-43. doi: 10.1101/gr.090597.108. Epub 2009 Jun 29. Genome Res. 2009. PMID: 19564452 Free PMC article.
-
ASPic-GeneID: a lightweight pipeline for gene prediction and alternative isoforms detection.Biomed Res Int. 2013;2013:502827. doi: 10.1155/2013/502827. Epub 2013 Nov 7. Biomed Res Int. 2013. PMID: 24308000 Free PMC article.
-
Whole-Genome Alignment and Comparative Annotation.Annu Rev Anim Biosci. 2019 Feb 15;7:41-64. doi: 10.1146/annurev-animal-020518-115005. Epub 2018 Oct 31. Annu Rev Anim Biosci. 2019. PMID: 30379572 Free PMC article. Review.
-
Simultaneous gene finding in multiple genomes.Bioinformatics. 2016 Nov 15;32(22):3388-3395. doi: 10.1093/bioinformatics/btw494. Epub 2016 Jul 27. Bioinformatics. 2016. PMID: 27466621 Free PMC article.
References
-
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. - PubMed
-
- Bafna V, Huson DH. The conserved exon method for gene finding. Proceedings of the Eighth International Conference on Intelligent Systems for Molecular Biology. 2000. pp. 3–12. - PubMed
-
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;17(Suppl 1):S140–S149. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
