

The properties of high-dimensional data spaces: implications for exploring gene and protein expression data
- PMID: 18097463
- PMCID: PMC2238676
- DOI: 10.1038/nrc2294
The properties of high-dimensional data spaces: implications for exploring gene and protein expression data
Abstract
High-throughput genomic and proteomic technologies are widely used in cancer research to build better predictive models of diagnosis, prognosis and therapy, to identify and characterize key signalling networks and to find new targets for drug development. These technologies present investigators with the task of extracting meaningful statistical and biological information from high-dimensional data spaces, wherein each sample is defined by hundreds or thousands of measurements, usually concurrently obtained. The properties of high dimensionality are often poorly understood or overlooked in data modelling and analysis. From the perspective of translational science, this Review discusses the properties of high-dimensional data spaces that arise in genomic and proteomic studies and the challenges they can pose for data analysis and interpretation.
Figures
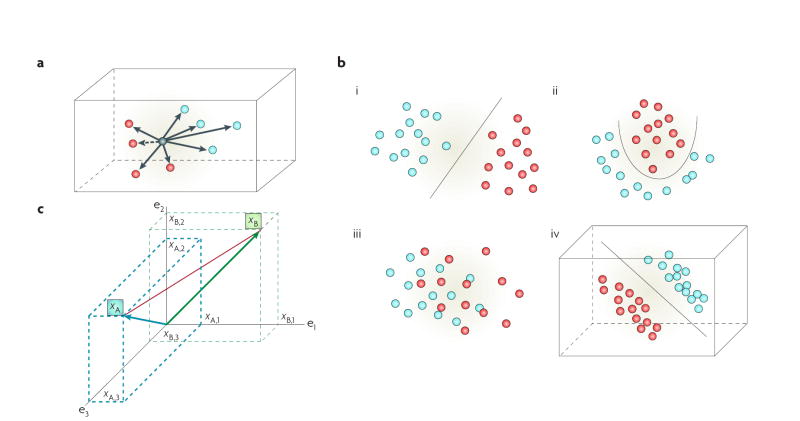
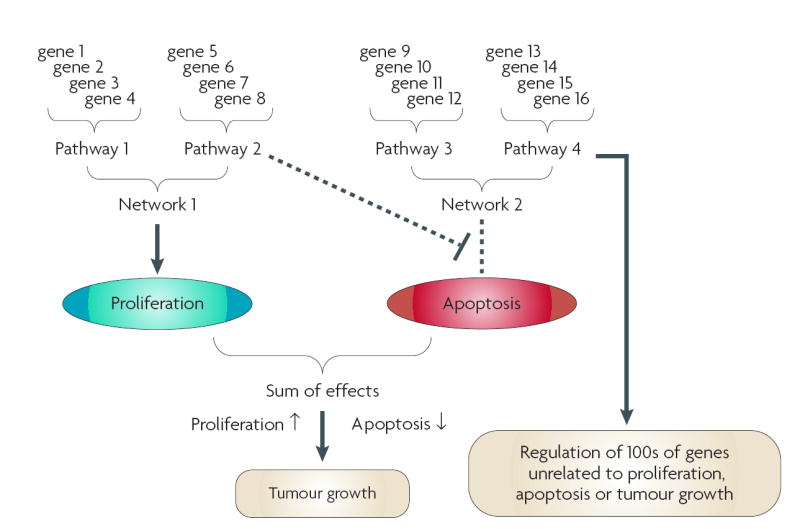
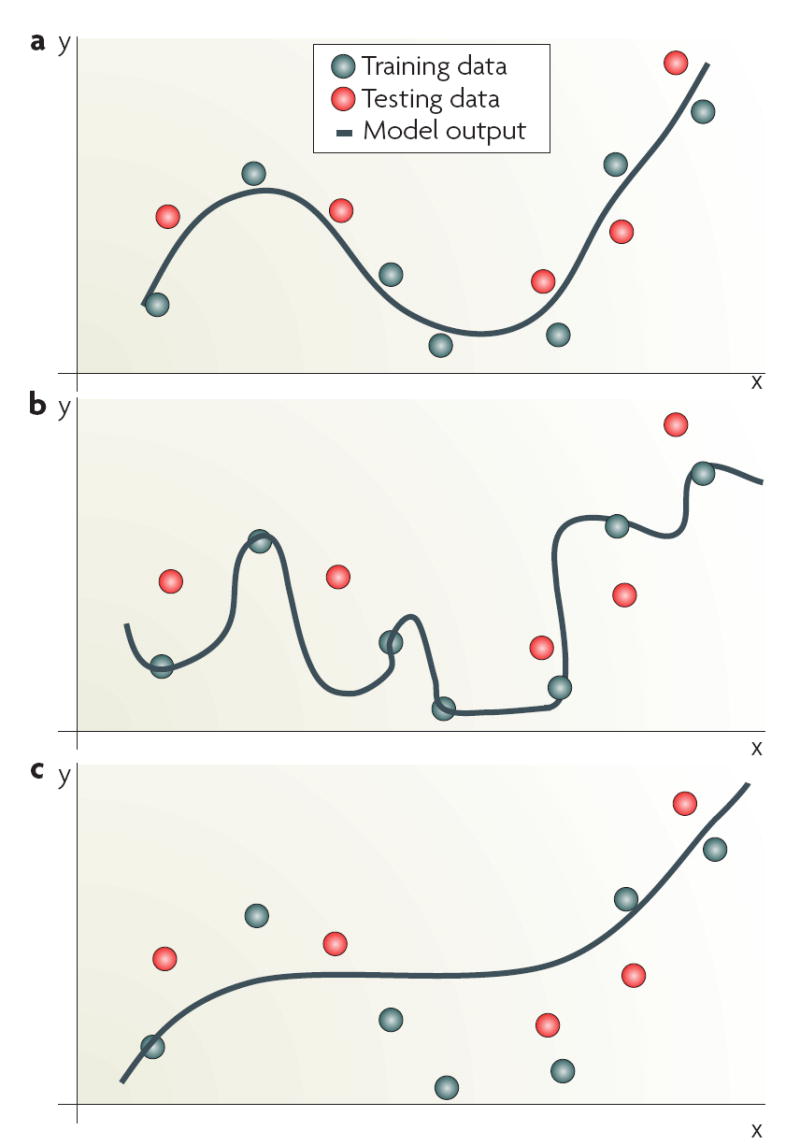
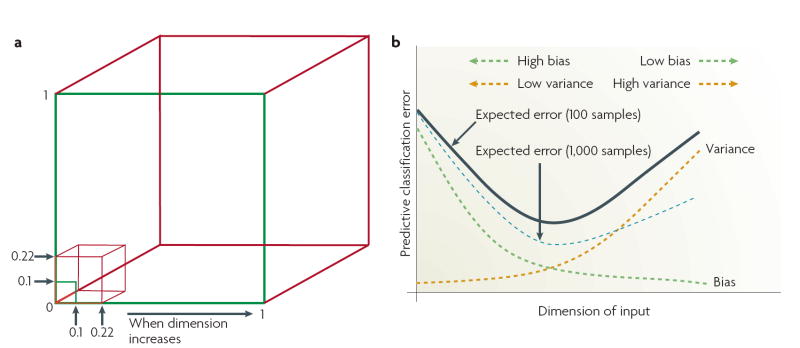
References
-
-
Khan J, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nature Med. 2001;7:673–679. Example of the successful use of molecular profiling to improve cancer diagnosis.
-
-
- Bhanot G, Alexe G, Levine AJ, Stolovitzky G. Robust diagnosis of non-Hodgkin lymphoma phenotypes validated on gene expression data from different laboratories. Genome Inform. 2005;16:233–244. - PubMed
-
- Lin YH, et al. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res. 2007;13:498–507. - PubMed
-
- Lopez-Rios F, et al. Global gene expression profiling of pleural mesotheliomas: overexpression of aurora kinases and P16/CDKN2A deletion as prognostic factors and critical evaluation of microarray-based prognostic prediction. Cancer Res. 2006;66:2970–2979. - PubMed
-
- Ganly I, et al. Identification of angiogenesis/ metastases genes predicting chemoradiotherapy response in patients with laryngopharyngeal carcinoma. J Clin Oncol. 2007;25:1369–1376. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
