

Bioconjugation of oligonucleotides for treating liver fibrosis
- PMID: 18154454
- PMCID: PMC2777659
- DOI: 10.1089/oli.2007.0097
Bioconjugation of oligonucleotides for treating liver fibrosis
Abstract
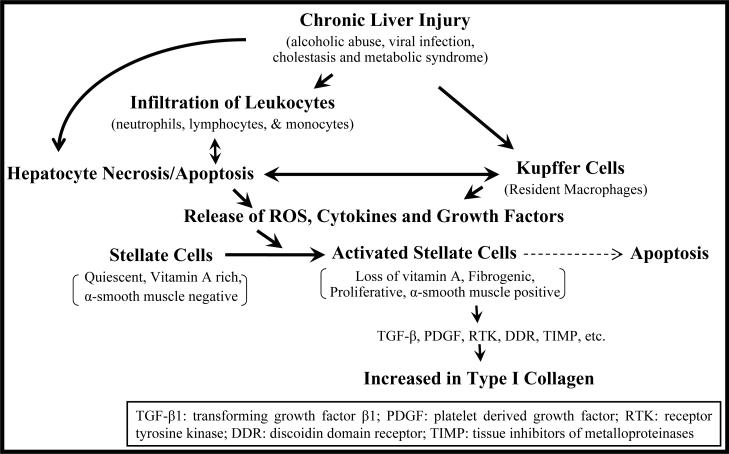
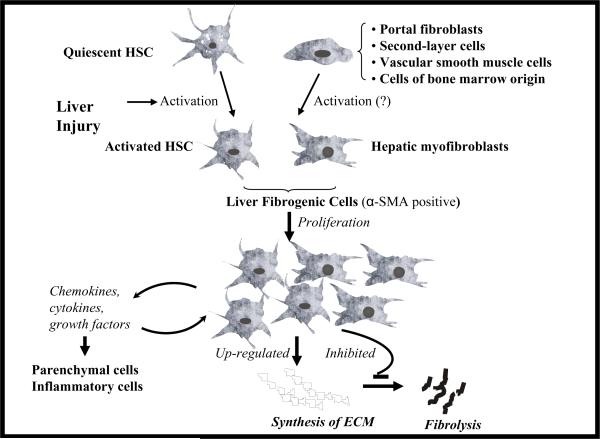
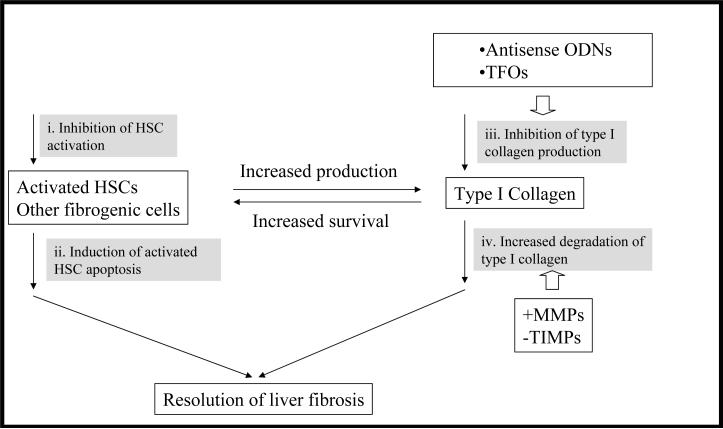
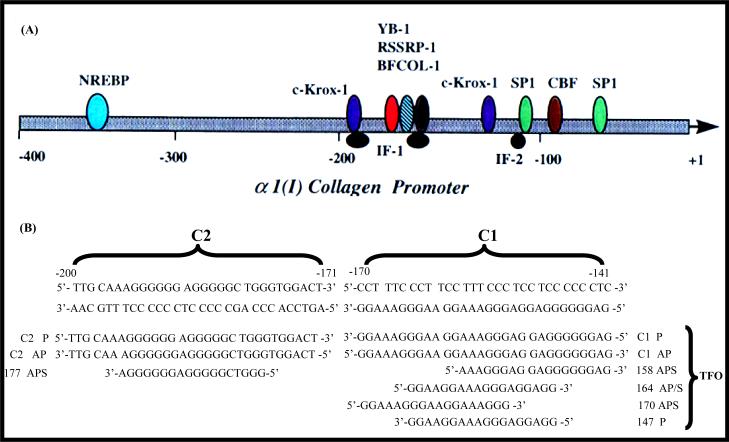
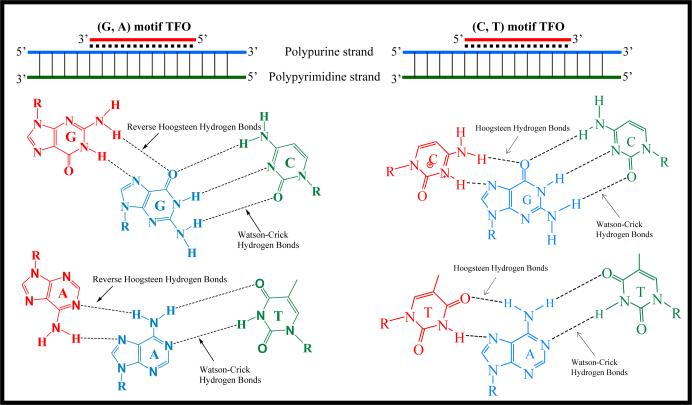
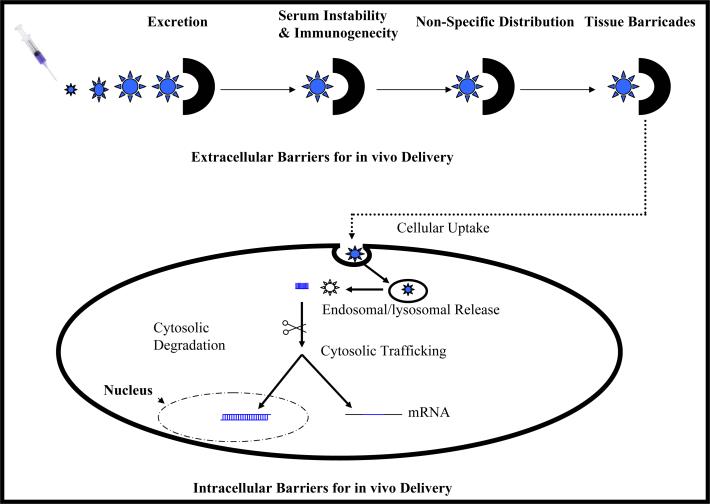
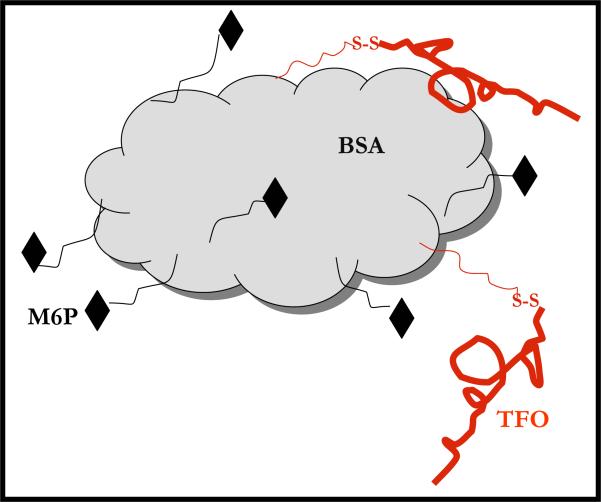
Liver fibrosis results from chronic liver injury due to hepatitis B and C, excessive alcohol ingestion, and metal ion overload. Fibrosis culminates in cirrhosis and results in liver failure. Therefore, a potent antifibrotic therapy is urgently needed to reverse scarring and eliminate progression to cirrhosis. Although activated hepatic stellate cells (HSCs) remain the principle cell type responsible for liver fibrosis, perivascular fibroblasts of portal and central veins as well as periductular fibroblasts are other sources of fibrogenic cells. This review will critically discuss various treatment strategies for liver fibrosis, including prevention of liver injury, reduction of inflammation, inhibition of HSC activation, degradation of scar matrix, and inhibition of aberrant collagen synthesis. Oligonucleotides (ODNs) are short, single-stranded nucleic acids, which disrupt expression of target protein by binding to complementary mRNA or forming triplex with genomic DNA. Triplex forming oligonucleotides (TFOs) provide an attractive strategy for treating liver fibrosis. A series of TFOs have been developed for inhibiting the transcription of alpha1(I) collagen gene, which opens a new area for antifibrotic drugs. There will be in-depth discussion on the use of TFOs and how different bioconjugation strategies can be utilized for their site-specific delivery to HSCs or hepatocytes for enhanced antifibrotic activities. Various insights developed in individual strategy and the need for multipronged approaches will also be discussed.
Figures

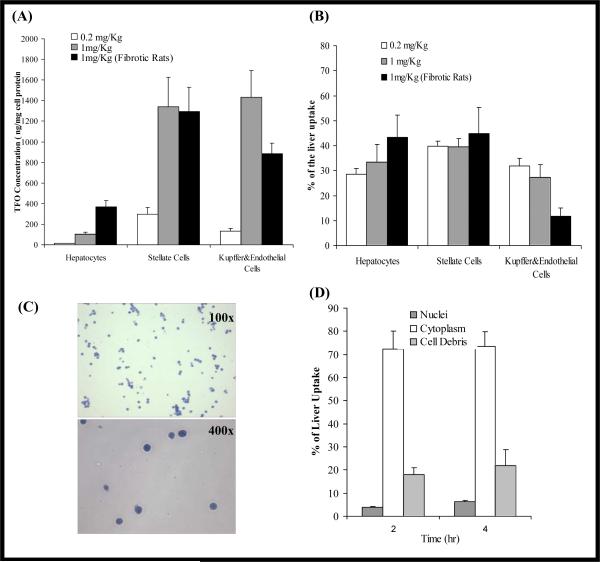
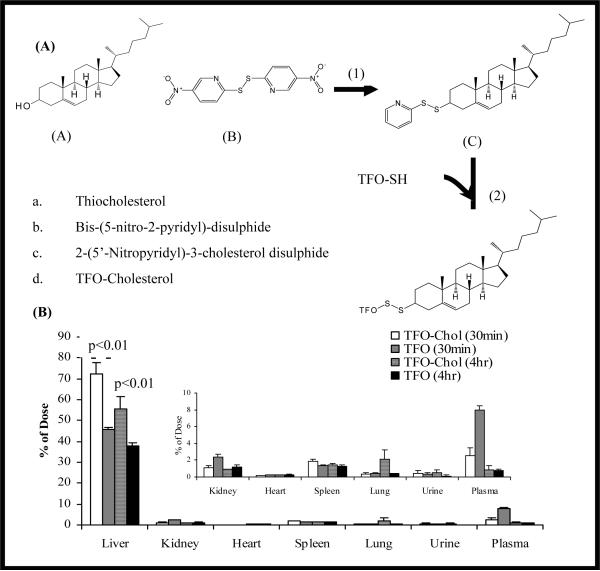
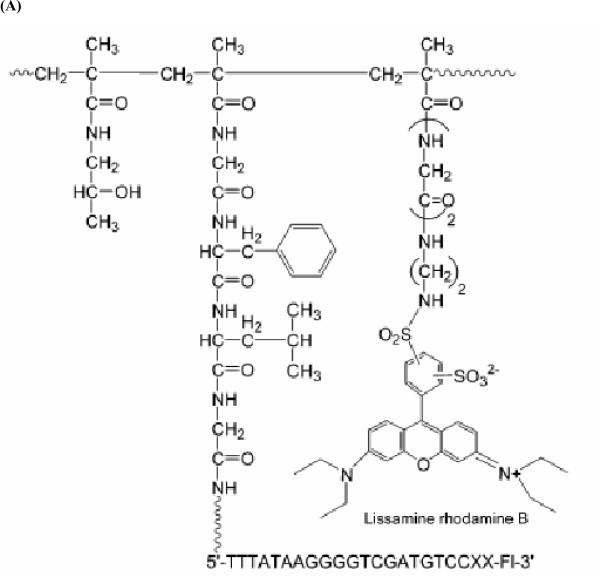
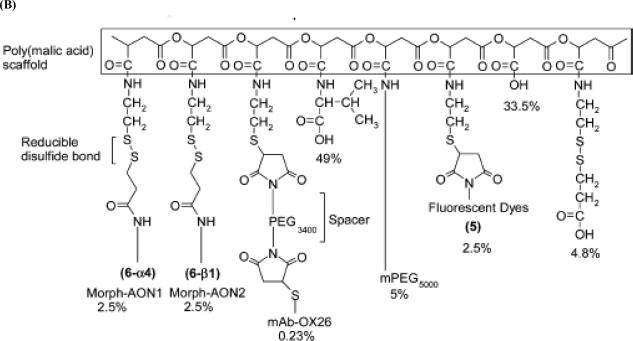
Similar articles
-
Gene modulation for treating liver fibrosis.Crit Rev Ther Drug Carrier Syst. 2007;24(2):93-146. doi: 10.1615/critrevtherdrugcarriersyst.v24.i2.10. Crit Rev Ther Drug Carrier Syst. 2007. PMID: 17725523 Free PMC article. Review.
-
Prevention of liver fibrosis by triple helix-forming oligodeoxyribonucleotides targeted to the promoter region of type I collagen gene.Oligonucleotides. 2010 Oct;20(5):231-7. doi: 10.1089/oli.2010.0244. Epub 2010 Sep 6. Oligonucleotides. 2010. PMID: 20818932 Free PMC article.
-
Fibromodulin, an oxidative stress-sensitive proteoglycan, regulates the fibrogenic response to liver injury in mice.Gastroenterology. 2012 Mar;142(3):612-621.e5. doi: 10.1053/j.gastro.2011.11.029. Epub 2011 Dec 1. Gastroenterology. 2012. PMID: 22138190 Free PMC article.
-
Collagenase Type I and Probucol-Loaded Nanoparticles Penetrate the Extracellular Matrix to Target Hepatic Stellate Cells for Hepatic Fibrosis Therapy.Acta Biomater. 2024 Feb;175:262-278. doi: 10.1016/j.actbio.2023.12.027. Epub 2023 Dec 21. Acta Biomater. 2024. PMID: 38141933
-
Targeted TFO delivery to hepatic stellate cells.J Control Release. 2011 Oct 30;155(2):326-30. doi: 10.1016/j.jconrel.2011.06.037. Epub 2011 Jul 8. J Control Release. 2011. PMID: 21763370 Free PMC article. Review.
Cited by
-
Prospective of extracellular matrix and drug correlations in disease management.Asian J Pharm Sci. 2021 Mar;16(2):147-160. doi: 10.1016/j.ajps.2020.06.007. Epub 2020 Aug 30. Asian J Pharm Sci. 2021. PMID: 33995610 Free PMC article. Review.
-
STAT3 Decoy Oligodeoxynucleotides Suppress Liver Inflammation and Fibrosis in Liver Cancer Cells and a DDC-Induced Liver Injury Mouse Model.Molecules. 2024 Jan 25;29(3):593. doi: 10.3390/molecules29030593. Molecules. 2024. PMID: 38338338 Free PMC article.
-
Small Leucine-Rich Proteoglycans in Skin Wound Healing.Front Pharmacol. 2020 Jan 28;10:1649. doi: 10.3389/fphar.2019.01649. eCollection 2019. Front Pharmacol. 2020. PMID: 32063855 Free PMC article. Review.
-
Liver-targeted gene therapy: Approaches and challenges.Liver Transpl. 2015 Jun;21(6):718-37. doi: 10.1002/lt.24122. Liver Transpl. 2015. PMID: 25824605 Free PMC article. Review.
-
Induction of heme oxygenase-1 protects against nutritional fibrosing steatohepatitis in mice.Lipids Health Dis. 2011 Feb 12;10:31. doi: 10.1186/1476-511X-10-31. Lipids Health Dis. 2011. PMID: 21314960 Free PMC article.
References
-
- Raghow R. The role of extracellular matrix in postinflammatory wound healing and fibrosis. Faseb J. 1994;8:823–31. - PubMed
-
- Anderson RN. Deaths: leading causes for 2000. Natl Vital Stat Rep. 2002;50:1–85. - PubMed
-
- Gines P, Cardenas A, Arroyo V, Rodes J. Management of cirrhosis and ascites. N Engl J Med. 2004;350:1646–54. - PubMed
-
- El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36:S74–83. - PubMed
-
- Befeler AS, Di Bisceglie AM. Hepatocellular carcinoma: diagnosis and treatment. Gastroenterology. 2002;122:1609–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
