

Crystal structure at 2.8 A of Huntingtin-interacting protein 1 (HIP1) coiled-coil domain reveals a charged surface suitable for HIP1 protein interactor (HIPPI)
- PMID: 18155047
- PMCID: PMC2271068
- DOI: 10.1016/j.jmb.2007.11.036
Crystal structure at 2.8 A of Huntingtin-interacting protein 1 (HIP1) coiled-coil domain reveals a charged surface suitable for HIP1 protein interactor (HIPPI)
Abstract
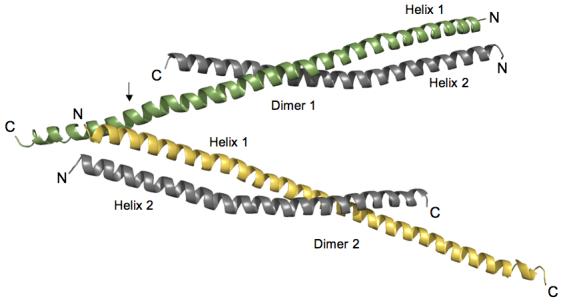
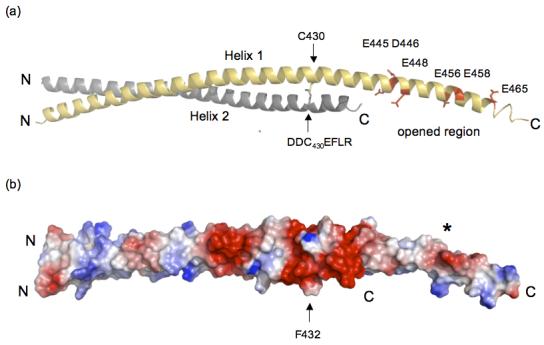
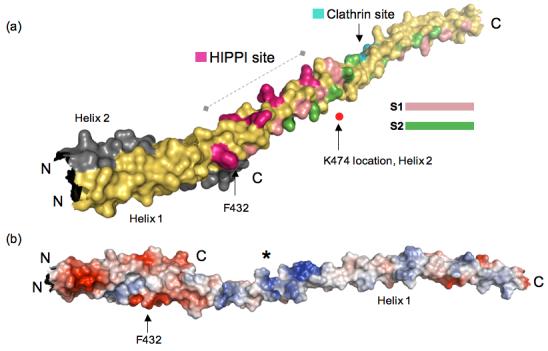
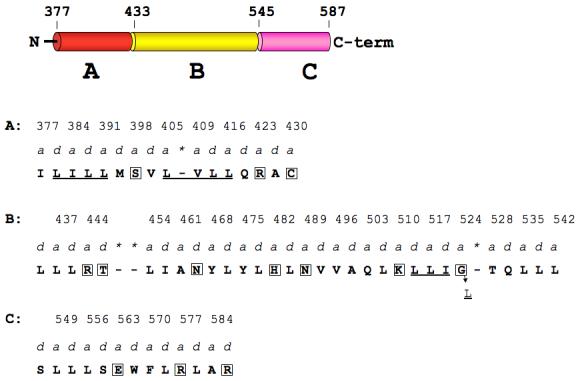
Huntington's disease is a genetic neurological disorder that is triggered by the dissociation of the huntingtin protein (htt) from its obligate interaction partner Huntingtin-interacting protein 1 (HIP1). The release of the huntingtin protein permits HIP1 protein interactor (HIPPI) to bind to its recognition site on HIP1 to form a HIPPI/HIP1 complex that recruits procaspase-8 to begin the process of apoptosis. The interaction module between HIPPI and HIP1 was predicted to resemble a death-effector domain. Our 2.8-A crystal structure of the HIP1 371-481 subfragment that includes F432 and K474, which is important for HIPPI binding, is not a death-effector domain but is a partially opened coiled coil. The HIP1 371-481 model reveals a basic surface that we hypothesize to be suitable for binding HIPPI. There is an opened region next to the putative HIPPI site that is highly negatively charged. The acidic residues in this region are highly conserved in HIP1 and a related protein, HIP1R, from different organisms but are not conserved in the yeast homologue of HIP1, sla2p. We have modeled approximately 85% of the coiled-coil domain by joining our new HIP1 371-481 structure to the HIP1 482-586 model (Protein Data Bank code: 2NO2). Finally, the middle of this coiled-coil domain may be intrinsically flexible and suggests a new interaction model where HIPPI binds to a U-shaped HIP1 molecule.
Figures
References
-
- Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. - PubMed
-
- Wanker EE, Rovira C, Scherzinger E, Hasenbank R, Walter S, Tait D, Colicelli J, Lehrach H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet. 1997;6:487–95. - PubMed
-
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst C-A, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the huntingtin-interacting protein HIP-1 and a novel partner HIPPI. Nature Cell Biol. 2002;4:95–105. - PubMed
-
- Chen C-Y, Brodsky FM. Huntington-interacting protein 1 (Hip1) and Hip1-related protein (Hip1R) bind the conserved sequence of clathrin light chains and thereby influence clathrin assembly in vitro and actin distribution in vivo. J. Biol. Chem. 2005;280:6109–6117. - PubMed
-
- Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J. Biol. Chem. 2001;276:46230–46236. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
