

Arrhythmogenic right ventricular cardiomyopathy/dysplasia: risk stratification and therapy
- PMID: 18156007
- PMCID: PMC2225487
- DOI: 10.1016/j.pcad.2007.10.004
Arrhythmogenic right ventricular cardiomyopathy/dysplasia: risk stratification and therapy
Abstract
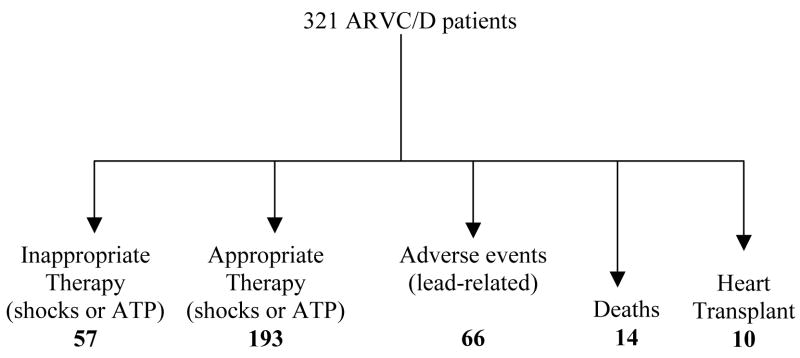
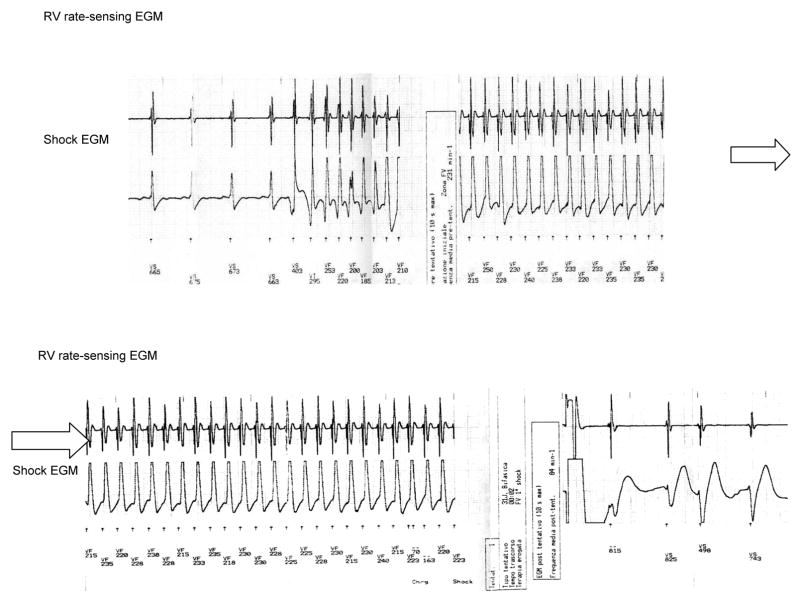
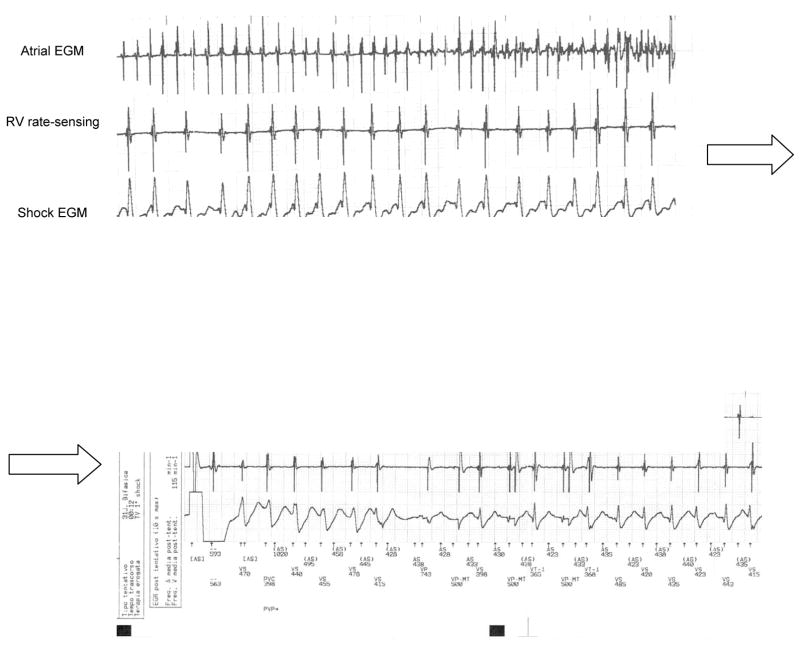
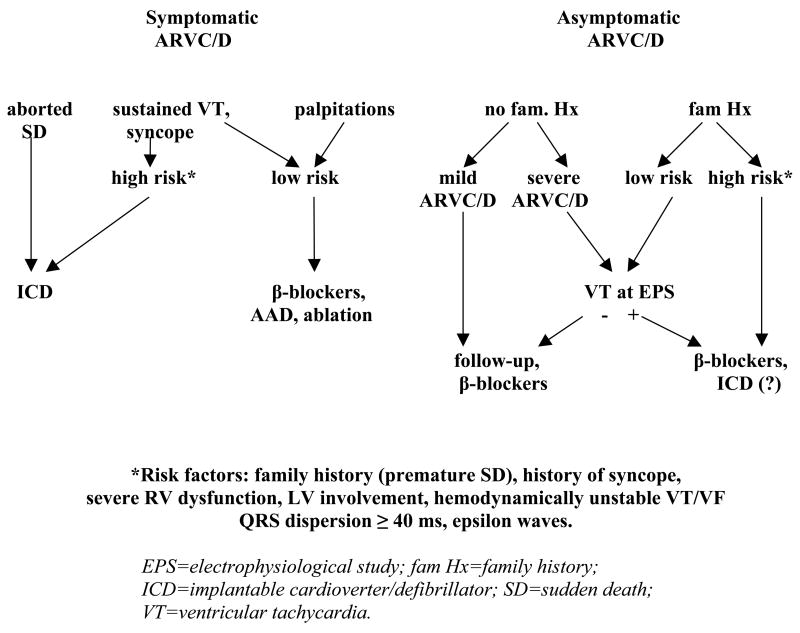
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is an inherited heart muscle disease that occurs primarily in young and middle-age individuals. It is characterized by ventricular arrhythmias (VA), sudden death, and by heart failure occurring later in life (–5). Ventricular electrical instability may occur at any time during the disease depending upon possible different pathophysiologic mechanisms including: a) Inflammation and apoptosis leading to ventricular fibrillation; or b) Fibro-fatty tissue repair leading to scar-related ventricular tachycardia (VT). Heart failure may occur later in life secondary to slow, progressive loss of right and left ventricular myocardium (–8). The role of pharmacological therapy in controlling VA and preventing sudden death has been evaluated in single-center studies (9), and the results of catheter ablation have been described in small series of patients (–15). The efficacy and safety of implantable cardioverter/defibrillator (ICD) have also been reported in small, single-center studies (–19) and recently in larger single and multicenter studies (–27). The main questions regarding the risk stratification and the therapeutic strategy in ARVC/D are: 1) differential diagnosis with idiopathic VT (right ventricular outflow tract VT) in an apparently normal heart. The prognosis of this latter condition is usually excellent with rare cases of sudden cardiac death; 2) prognostic and therapeutic significance of noninvasive and invasive investigations including electrophysiologic study (EPS); 3) efficacy of antiarrhythmic drugs (AAD) to prevent VT and sudden cardiac death and the adverse effects of these drugs in this population; 4) indications and results of catheter ablation; 5) identification of patients at high risk of sudden cardiac death who need ICD implantation as well as the complications of ICD in a diseased right ventricular myocardium. It is important to recognize that ARVC/D is a progressive disease and risk factors may change during follow-up requiring periodic revaluation of risk as well as of therapy.
With the identification of family members who carry a disease causing gene, the therapeutic dilemma has broadened to include risk stratification in genotype positive family members who may have occasional ventricular ectopy or have no clinical evidence of the disease.
Figures
References
-
- Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–98. - PubMed
-
- Thiene G, Nava A, Corrado D, et al. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33. - PubMed
-
- Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–91. - PubMed
-
- Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–20. - PubMed
-
- Fontaine GH, Fontaliran F, Frank R. Arrhythmogenic right ventricular cardiomyopathy: clinical forms and main differential diagnoses. Circulation. 1998;97:1532–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
