

Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations
- PMID: 18158359
- PMCID: PMC2957811
- DOI: 10.1161/CIRCULATIONAHA.107.726752
Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations
Abstract
Background: PRKAG2 mutations cause glycogen-storage cardiomyopathy, ventricular preexcitation, and conduction system degeneration. A genetic approach that utilizes a binary inducible transgenic system was used to investigate the disease mechanism and to assess preventability and reversibility of disease features in a mouse model of glycogen-storage cardiomyopathy.
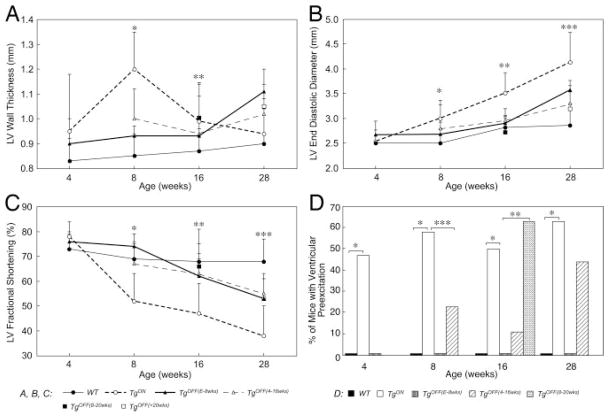
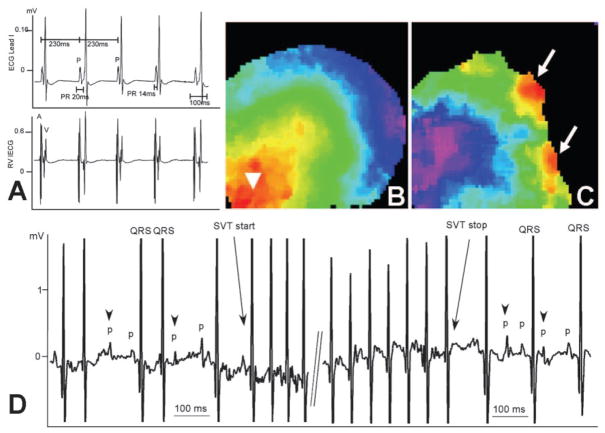
Methods and results: Transgenic (Tg) mice expressing a human N488I PRKAG2 cDNA under control of the tetracycline-repressible alpha-myosin heavy chain promoter underwent echocardiography, ECG, and in vivo electrophysiology studies. Transgene suppression by tetracycline administration caused a reduction in cardiac glycogen content and was initiated either prenatally (Tg(OFF(E-8 weeks))) or at different time points during life (Tg(OFF(4-16 weeks)), Tg(OFF(8-20 weeks)), and Tg(OFF(>20 weeks))). One group never received tetracycline, expressing transgene throughout life (Tg(ON)). Tg(ON) mice developed cardiac hypertrophy followed by dilatation, ventricular preexcitation involving multiple accessory pathways, and conduction system disease, including sinus and atrioventricular node dysfunction.
Conclusions: Using an externally modifiable transgenic system, cardiomyopathy, cardiac dysfunction, and electrophysiological disorders were demonstrated to be reversible processes in PRKAG2 disease. Transgene suppression during early postnatal development prevented the development of accessory electrical pathways but not cardiomyopathy or conduction system degeneration. Taken together, these data provide insight into mechanisms of cardiac PRKAG2 disease and suggest that glycogen-storage cardiomyopathy can be modulated by lowering glycogen content in the heart.
Figures




References
-
- Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10:1215–1220. - PubMed
-
- Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–488. - PubMed
-
- Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology. 2006;21:48–60. - PubMed
-
- Ingwall JS. Transgenesis and cardiac energetics: new insights into cardiac metabolism. J Mol Cell Cardiol. 2004;37:613–623. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
