

Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD
- PMID: 18171917
- PMCID: PMC2486412
- DOI: 10.1523/JNEUROSCI.4405-07.2008
Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD
Abstract
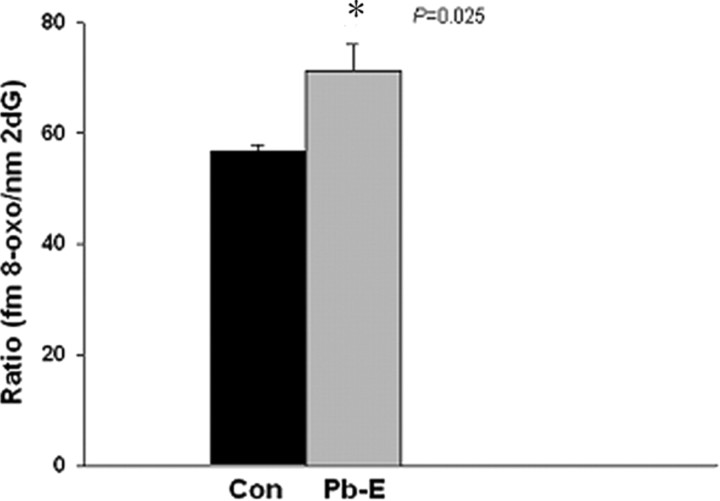
The sporadic nature of Alzheimer's disease (AD) argues for an environmental link that may drive AD pathogenesis; however, the triggering factors and the period of their action are unknown. Recent studies in rodents have shown that exposure to lead (Pb) during brain development predetermined the expression and regulation of the amyloid precursor protein (APP) and its amyloidogenic beta-amyloid (Abeta) product in old age. Here, we report that the expression of AD-related genes [APP, BACE1 (beta-site APP cleaving enzyme 1)] as well as their transcriptional regulator (Sp1) were elevated in aged (23-year-old) monkeys exposed to Pb as infants. Furthermore, developmental exposure to Pb altered the levels, characteristics, and intracellular distribution of Abeta staining and amyloid plaques in the frontal association cortex. These latent effects were accompanied by a decrease in DNA methyltransferase activity and higher levels of oxidative damage to DNA, indicating that epigenetic imprinting in early life influenced the expression of AD-related genes and promoted DNA damage and pathogenesis. These data suggest that AD pathogenesis is influenced by early life exposures and argue for both an environmental trigger and a developmental origin of AD.
Figures





References
-
- Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, Tsuang MT. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet. 2004;127:51–59. - PubMed
-
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134:60–66. - PubMed
-
- Ball MJ, Lo P. Granulovacuolar degeneration in the ageing brain and in dementia. J Neuropathol Exp Neurol. 1977;36:474–487. - PubMed
-
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- AG027246/AG/NIA NIH HHS/United States
- R01 AG018884/AG/NIA NIH HHS/United States
- R0AG18884/AG/NIA NIH HHS/United States
- Z01 ES021164/ImNIH/Intramural NIH HHS/United States
- ES013022/ES/NIEHS NIH HHS/United States
- R21 ES013022/ES/NIEHS NIH HHS/United States
- P20 RR016457/RR/NCRR NIH HHS/United States
- R0AG18379/AG/NIA NIH HHS/United States
- R15 AG023604/AG/NIA NIH HHS/United States
- R01 AG018379/AG/NIA NIH HHS/United States
- P20RR016457/RR/NCRR NIH HHS/United States
- R03 AG027246/AG/NIA NIH HHS/United States
- 1R15AG023604-01/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases