

Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling
- PMID: 18172167
- PMCID: PMC2151009
- DOI: 10.1101/gad.1590908
Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling
Abstract
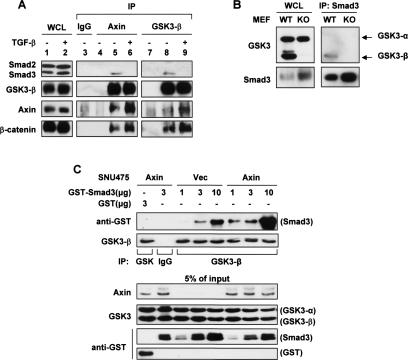
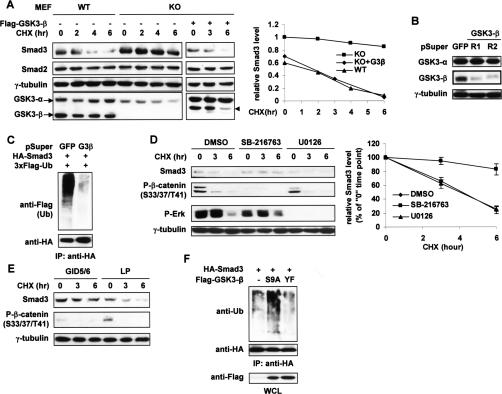
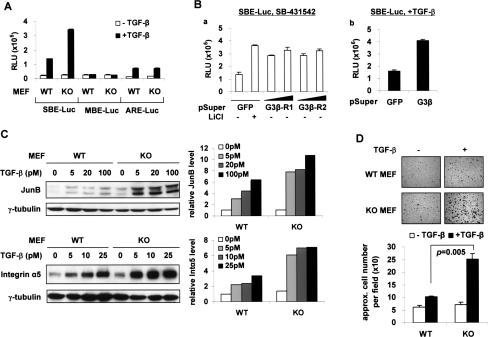
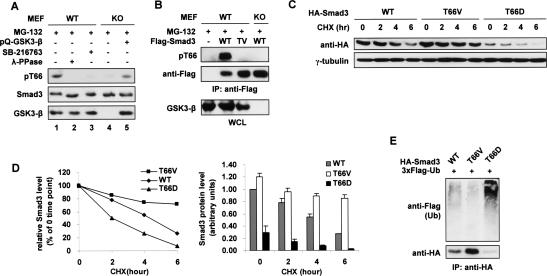
The broad range of biological responses elicited by transforming growth factor-beta (TGF-beta) in various types of tissues and cells is mainly determined by the expression level and activity of the effector proteins Smad2 and Smad3. It is not fully understood how the baseline properties of Smad3 are regulated, although this molecule is in complex with many other proteins at the steady state. Here we show that nonactivated Smad3, but not Smad2, undergoes proteasome-dependent degradation due to the concerted action of the scaffolding protein Axin and its associated kinase, glycogen synthase kinase 3-beta (GSK3-beta). Smad3 physically interacts with Axin and GSK3-beta only in the absence of TGF-beta. Reduction in the expression or activity of Axin/GSK3-beta leads to increased Smad3 stability and transcriptional activity without affecting TGF-beta receptors or Smad2, whereas overexpression of these proteins promotes Smad3 basal degradation and desensitizes cells to TGF-beta. Mechanistically, Axin facilitates GSK3-beta-mediated phosphorylation of Smad3 at Thr66, which triggers Smad3 ubiquitination and degradation. Thr66 mutants of Smad3 show altered protein stability and hence transcriptional activity. These results indicate that the steady-state stability of Smad3 is an important determinant of cellular sensitivity to TGF-beta, and suggest a new function of the Axin/GSK3-beta complex in modulating critical TGF-beta/Smad3-regulated processes during development and tumor progression.
Figures



References
-
- Bierie B., Moses H.L. Tumour microenvironment: TGFβ: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer. 2006;6:506–520. - PubMed
-
- Clarke D.C., Betterton M.D., Liu X. Systems theory of Smad signalling. IEE Proc. Syst. Biol. 2006;153:412–424. - PubMed
-
- Cohen P., Frame S. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001;2:769–776. - PubMed
-
- Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases