

Holoprosencephaly-Polydactyly syndrome: in search of an etiology
- PMID: 18178536
- PMCID: PMC2441840
- DOI: 10.1016/j.ejmg.2007.08.004
Holoprosencephaly-Polydactyly syndrome: in search of an etiology
Abstract
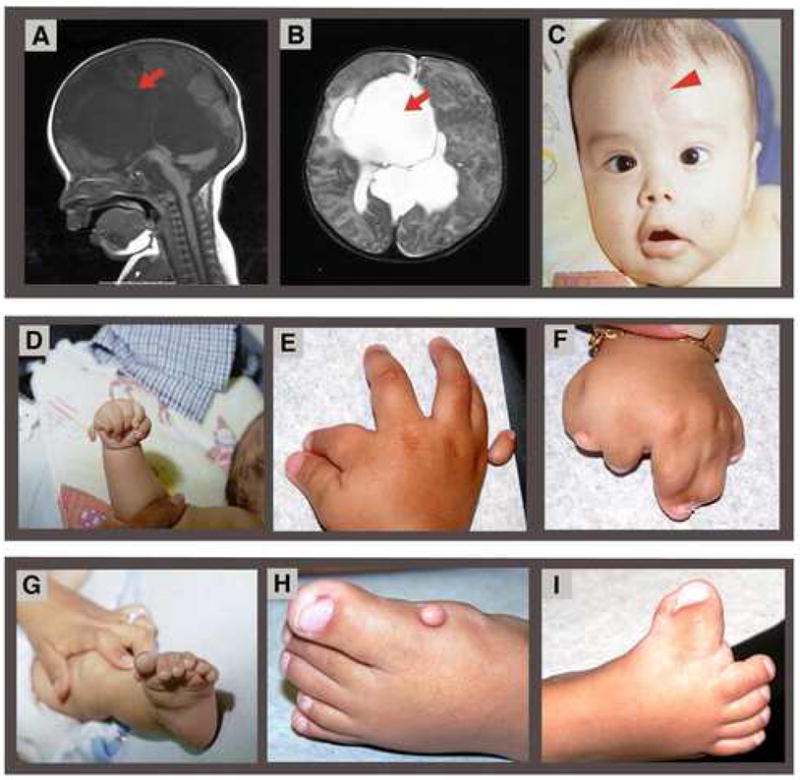
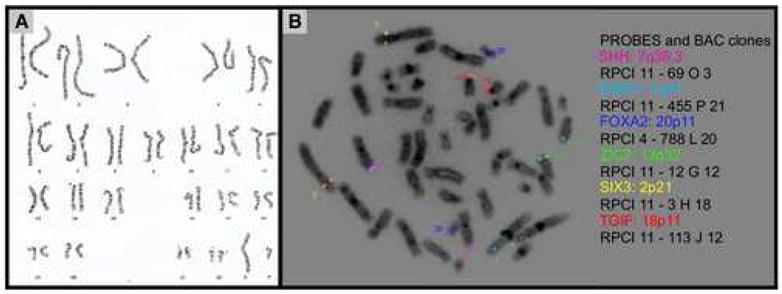
Holoprosencephaly-Polydactyly (HPS) or Pseudotrisomy 13 syndrome are names conferred to clinically categorize patients whose phenotype is congruent with Trisomy 13 in the context of a normal karyotype. The literature suggests that this entity may be secondary to submicroscopic deletions in holoprosencephaly (HPE) genes; however, a limited number of investigations have been undertaken to evaluate this hypothesis. To test this hypothesis we studied a patient with HPE, polydactyly, and craniofacial dysmorphologies consistent with the diagnosis of Trisomy 13 whose karyotype was normal. We performed mutational analysis in the four main HPE causing genes (SHH, SIX3, TGIF, and ZIC2) and GLI3, a gene associated with polydactyly as well as fluorescent in situ hybridization (FISH) to search for microdeletions in these genes and two candidate HPE genes (DISP1 and FOXA2). No mutations or deletions were detected. A whole genome approach utilizing array Comparative Genomic Hybridization (aCGH) to screen for copy number abnormalities was then taken. No loss or gain of DNA was noted. Although a single case, our results suggest that coding mutations in these HPE genes and copy number anomalies may not be causative in this disorder. Instead, HPS likely involves mutations in other genes integral in embryonic development of the forebrain, face and limbs. Our systematic analysis sets the framework to study other affected children and delineate the molecular etiology of this disorder.
Figures

References
-
- Bendavid C, Dubourg C, Gicquel I, Pasquier L, Saugier-Veber P, Durou MR, Jaillard S, Frebourg T, Haddad BR, Henry C, Odent S, David V. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2006;119:1–8. - PubMed
-
- Bendavid C, Haddad BR, Griffin A, Huizing M, Dubourg C, Gicquel I, Cavalli LR, Pasquier L, Shanske AL, Long R, Ouspenskaia M, Odent S, Lacbawan F, David V, Muenke M. Multicolour FISH and quantitative PCR can detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet. 2006;43:496–500. - PMC - PubMed
-
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–3. - PubMed
-
- Cohen MM, Jr, Gorlin RJ. Pseudo-trisomy 13 syndrome. Am J Med Genet. 1991;39:332–5. discussion 336–7. - PubMed
-
- Cohen MM, Jr, Shiota K. Teratogenesis of holoprosencephaly. Am J Med Genet. 2002;109:1–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
