

Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect
- PMID: 18178576
- PMCID: PMC2835541
- DOI: 10.1093/hmg/ddm379
Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect
Abstract
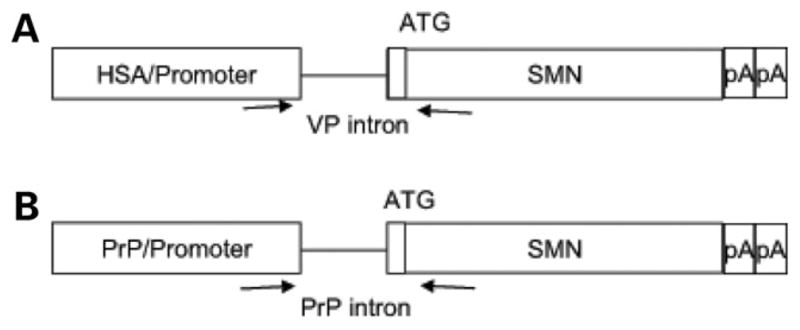
Spinal muscular atrophy (SMA) is caused by loss of the survival motor neuron gene (SMN1) and retention of the SMN2 gene. The copy number of SMN2 affects the amount of SMN protein produced and the severity of the SMA phenotype. While loss of mouse Smn is embryonic lethal, two copies of SMN2 prevents this embryonic lethality resulting in a mouse with severe SMA that dies 5 days after birth. Here we show that expression of full-length SMN under the prion promoter (PrP) rescues severe SMA mice. The PrP results in high levels of SMN in neurons at embryonic day 15. Mice homozygous for PrP-SMN with two copies of SMN2 and lacking mouse Smn survive for an average of 210 days and lumbar motor neuron root counts in these mice were normal. Expression of SMN solely in skeletal muscle using the human skeletal actin (HSA) promoter resulted in no improvement of the SMA phenotype or extension of survival. One HSA line displaying nerve expression of SMN did affect the SMA phenotype with mice living for an average of 160 days. Thus, we conclude that expression of full-length SMN in neurons can correct the severe SMA phenotype in mice. Furthermore, a small increase of SMN in neurons has a substantial impact on survival of SMA mice while high SMN levels in mature skeletal muscle alone has no impact.
Conflict of interest statement
Figures






References
-
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. - PubMed
-
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
