

Viral genome sequencing by random priming methods
- PMID: 18179705
- PMCID: PMC2254600
- DOI: 10.1186/1471-2164-9-5
Viral genome sequencing by random priming methods
Abstract
Background: Most emerging health threats are of zoonotic origin. For the overwhelming majority, their causative agents are RNA viruses which include but are not limited to HIV, Influenza, SARS, Ebola, Dengue, and Hantavirus. Of increasing importance therefore is a better understanding of global viral diversity to enable better surveillance and prediction of pandemic threats; this will require rapid and flexible methods for complete viral genome sequencing.
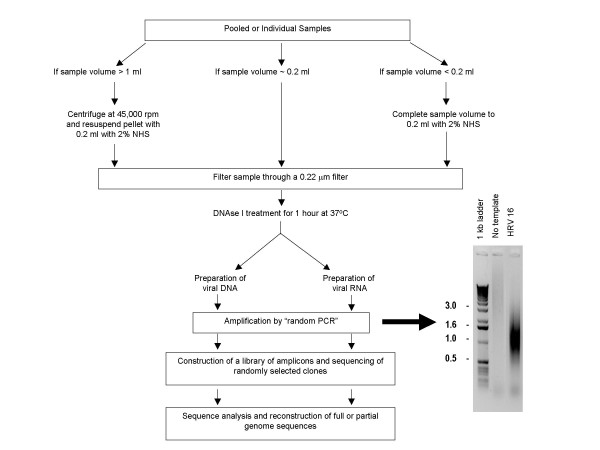
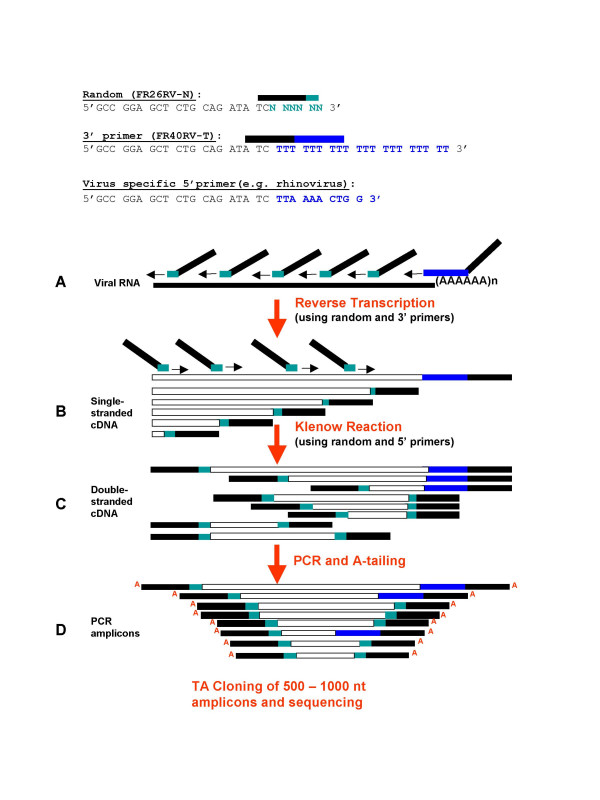
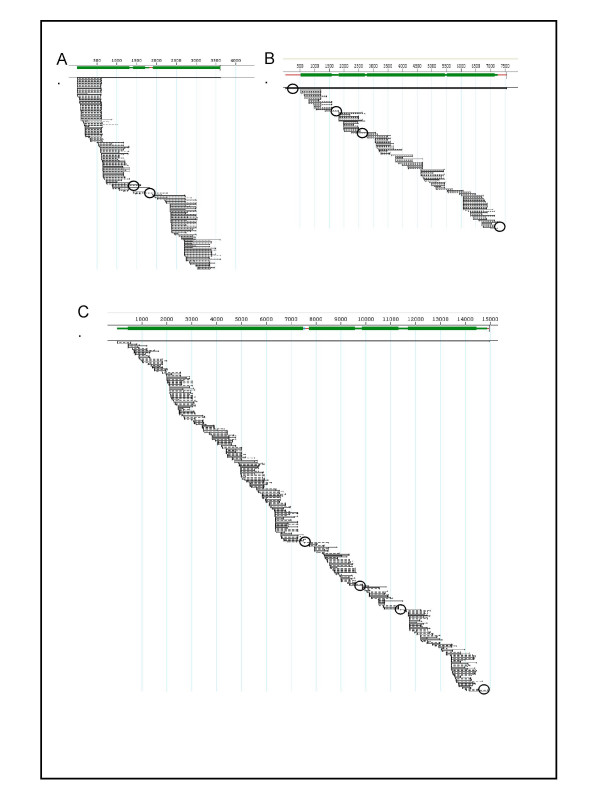
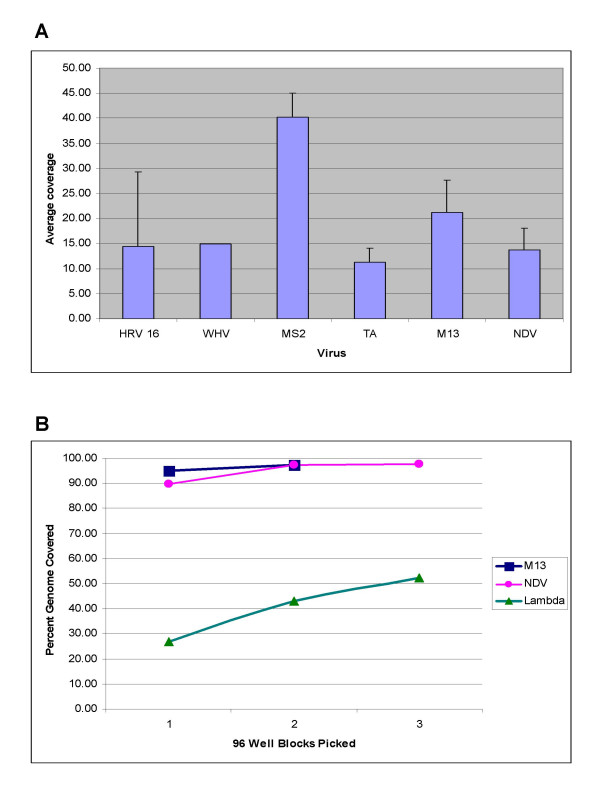
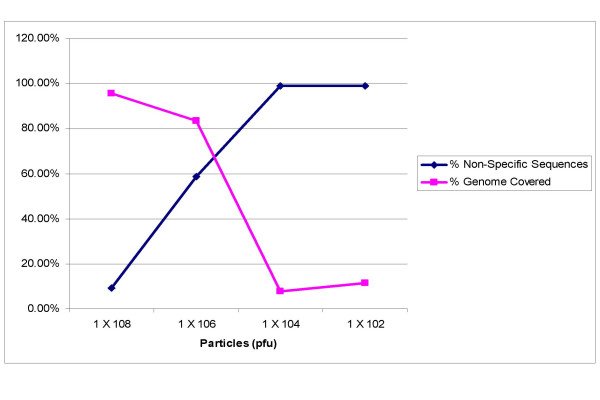
Results: We have adapted the SISPA methodology 123 to genome sequencing of RNA and DNA viruses. We have demonstrated the utility of the method on various types and sources of viruses, obtaining near complete genome sequence of viruses ranging in size from 3,000-15,000 kb with a median depth of coverage of 14.33. We used this technique to generate full viral genome sequence in the presence of host contaminants, using viral preparations from cell culture supernatant, allantoic fluid and fecal matter.
Conclusion: The method described is of great utility in generating whole genome assemblies for viruses with little or no available sequence information, viruses from greatly divergent families, previously uncharacterized viruses, or to more fully describe mixed viral infections.
Figures
References
-
- Allander T, Emerson SU, Engle RE, Purcell RH, Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11609–11614. doi: 10.1073/pnas.211424698. - DOI - PMC - PubMed
-
- Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12891–12896. doi: 10.1073/pnas.0504666102. - DOI - PMC - PubMed
-
- Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, Chan AM, Haynes M, Kelley S, Liu H, Mahaffy JM, Mueller JE, Nulton J, Olson R, Parsons R, Rayhawk S, Suttle CA, Rohwer F. The marine viromes of four oceanic regions. PLoS biology. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. - DOI - PMC - PubMed
-
- Breitbart M, Rohwer F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. BioTechniques. 2005;39:729–736. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
