

Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms
- PMID: 18179889
- PMCID: PMC2253956
- DOI: 10.1016/j.ajhg.2007.09.006
Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms
Abstract
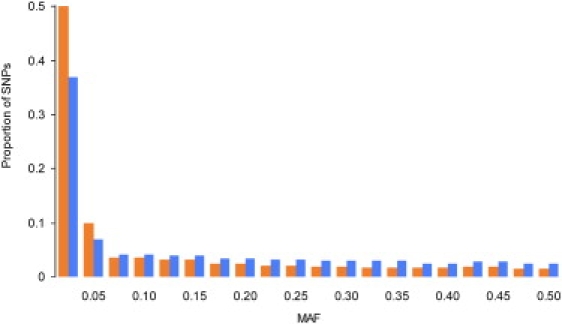
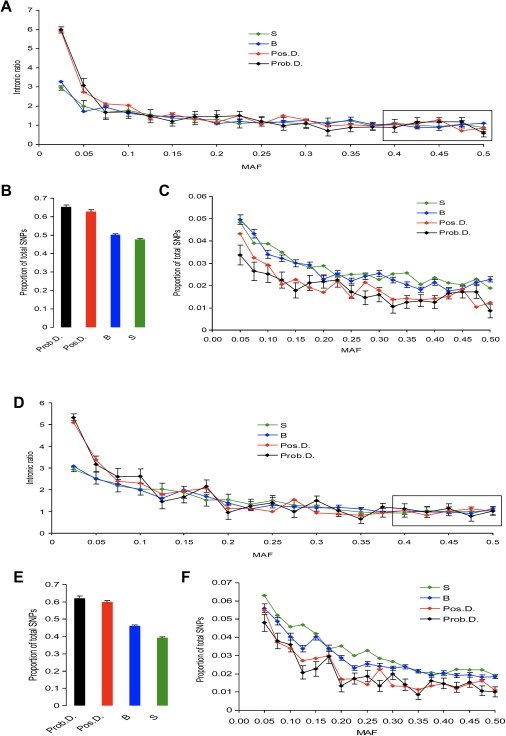
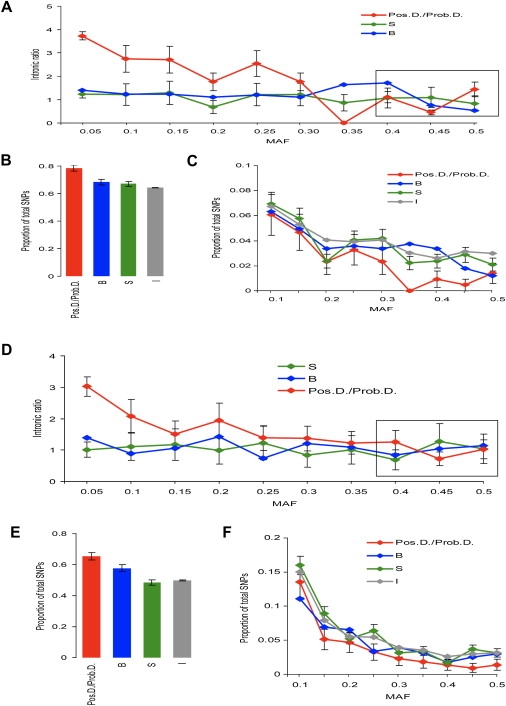
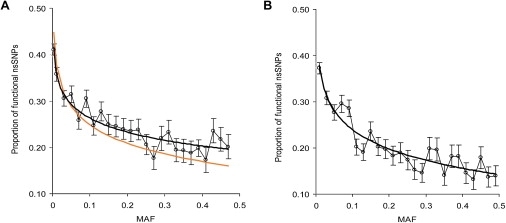
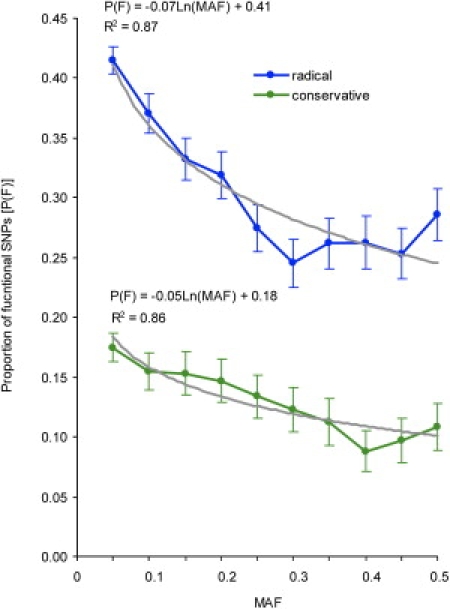
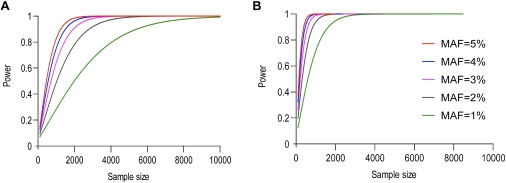
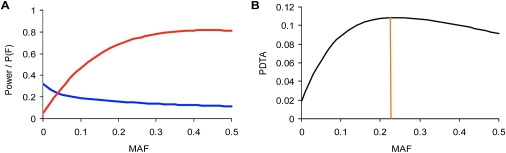
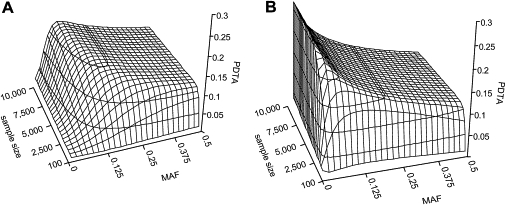
Currently, single-nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) of >5% are preferentially used in case-control association studies of common human diseases. Recent technological developments enable inexpensive and accurate genotyping of a large number of SNPs in thousands of cases and controls, which can provide adequate statistical power to analyze SNPs with MAF <5%. Our purpose was to determine whether evaluating rare SNPs in case-control association studies could help identify causal SNPs for common diseases. We suggest that slightly deleterious SNPs (sdSNPs) subjected to weak purifying selection are major players in genetic control of susceptibility to common diseases. We compared the distribution of MAFs of synonymous SNPs with that of nonsynonymous SNPs (1) predicted to be benign, (2) predicted to be possibly damaging, and (3) predicted to be probably damaging by PolyPhen. Our sources of data were the International HapMap Project, ENCODE, and the SeattleSNPs project. We found that the MAF distribution of possibly and probably damaging SNPs was shifted toward rare SNPs compared with the MAF distribution of benign and synonymous SNPs that are not likely to be functional. We also found an inverse relationship between MAF and the proportion of nsSNPs predicted to be protein disturbing. On the basis of this relationship, we estimated the joint probability that a SNP is functional and would be detected as significant in a case-control study. Our analysis suggests that including rare SNPs in genotyping platforms will advance identification of causal SNPs in case-control association studies, particularly as sample sizes increase.
Figures
References
-
- Risch N., Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. - PubMed
-
- Muller-Myhsok B., Abel L. Genetic analysis of complex diseases. Science. 1997;275:1328–1329. - PubMed
-
- Scott W.K., Pericak-Vance M.A., Haines J.L. Genetic analysis of complex diseases. Science. 1997;275:1327. - PubMed
-
- Long A.D., Grote M.N., Langley C.H. Genetic analysis of complex diseases. Science. 1997;275:1328. - PubMed
-
- The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
