

Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration
- PMID: 18179896
- PMCID: PMC2253967
- DOI: 10.1016/j.ajhg.2007.08.002
Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration
Abstract
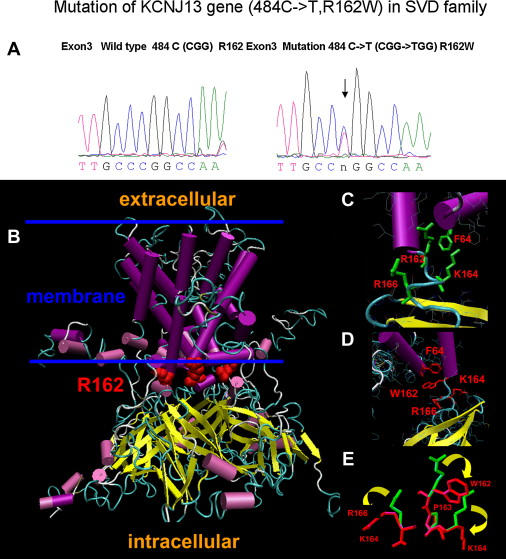
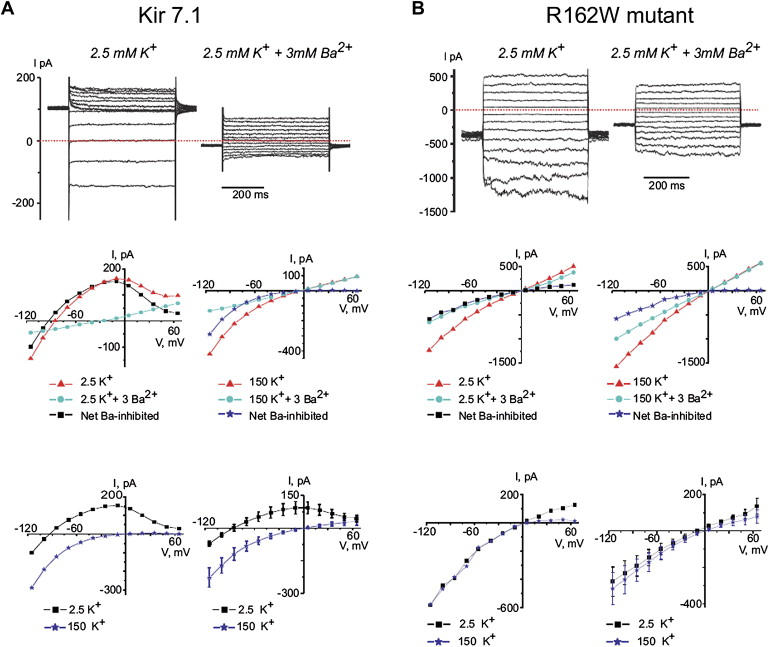
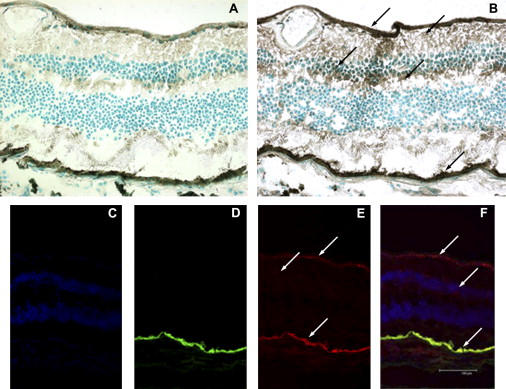
Snowflake vitreoretinal degeneration (SVD, MIM 193230) is a developmental and progressive hereditary eye disorder that affects multiple tissues within the eye. Diagnostic features of SVD include fibrillar degeneration of the vitreous humor, early-onset cataract, minute crystalline deposits in the neurosensory retina, and retinal detachment. A genome-wide scan previously localized the genetic locus for SVD to a 20 Mb region flanked by D2S2158 and D2S2202. This region contains 59 genes, of which 20 were sequenced, disclosing a heterozygous mutation (484C > T, R162W) in KCNJ13, member 13 of subfamily J of the potassium inwardly rectifying channel family in all affected individuals. The mutation in KCNJ13, the gene encoding Kir7.1, was not present in unaffected family members and 210 control individuals. Kir7.1 localized to human retina and retinal pigment epithelium and was especially prevalent in the internal limiting membrane adjacent to the vitreous body. Molecular modeling of this mutation predicted disruption of the structure of the potassium channel in the closed state located immediately adjacent to the cell-membrane inner boundary. Functionally, unlike wild-type Kir7.1 whose overexpression in CHO-K1 cells line produces highly selective potassium current, overexpression of R162W mutant Kir7.1 produces a nonselective cation current that depolarizes transfected cells and increases their fragility. These results indicate that the KCNJ13 R162W mutation can cause SVD and further show that vitreoretinal degeneration can arise through mutations in genes whose products are not structural components of the vitreous.
Figures
References
-
- Edwards A.O., Robertson J.E. Hereditary vitreoretinal degenerations. In: Ryan S.J., Hinton D.R., Schachat A.P., Wilkenson P., editors. Retina. Elsevier Inc.; Philadelphia: 2006. pp. 519–538.
-
- Lee M.M., Ritter R., III, Hirose T., Vu C.D., Edwards A.O. Snowflake vitreoretinal degeneration: Follow-up of the original family. Ophthalmology. 2003;110:2418–2426. - PubMed
-
- Hirose T., Lee K.Y., Schepens C.L. Snowflake degeneration in hereditary vitreoretinal degeneration. Am. J. Ophthalmol. 1974;77:143–153. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
