

A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor
- PMID: 18180305
- PMCID: PMC4228940
- DOI: 10.1074/jbc.M702881200
A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor
Abstract
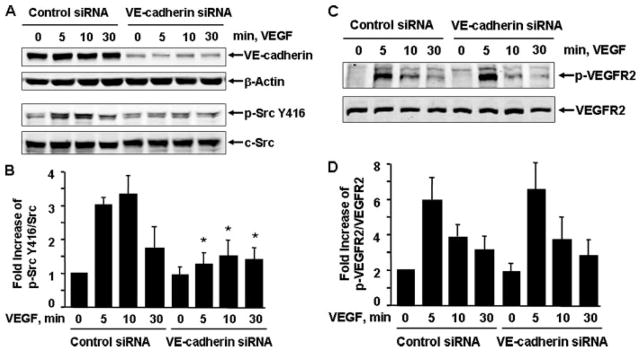
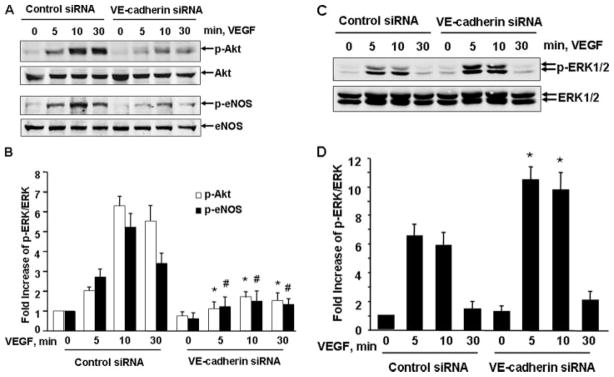
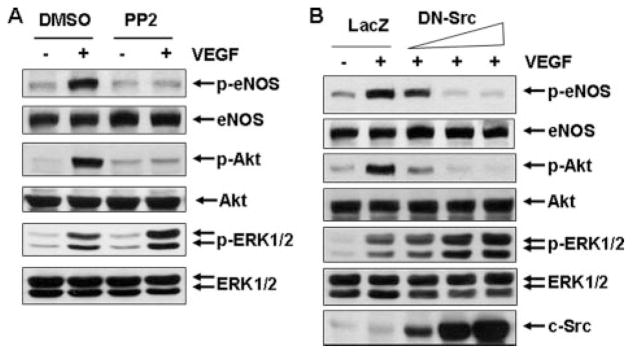
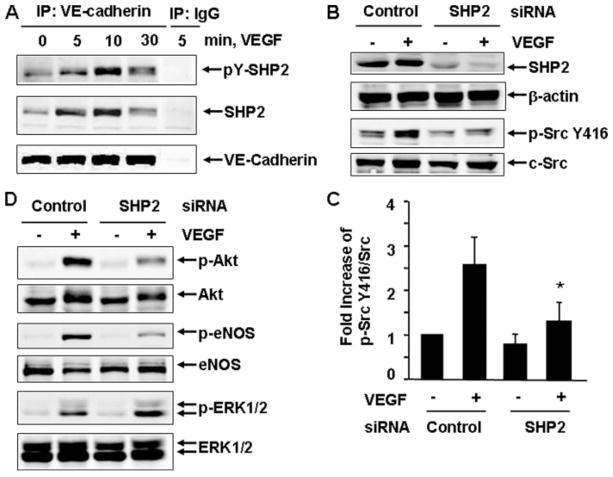
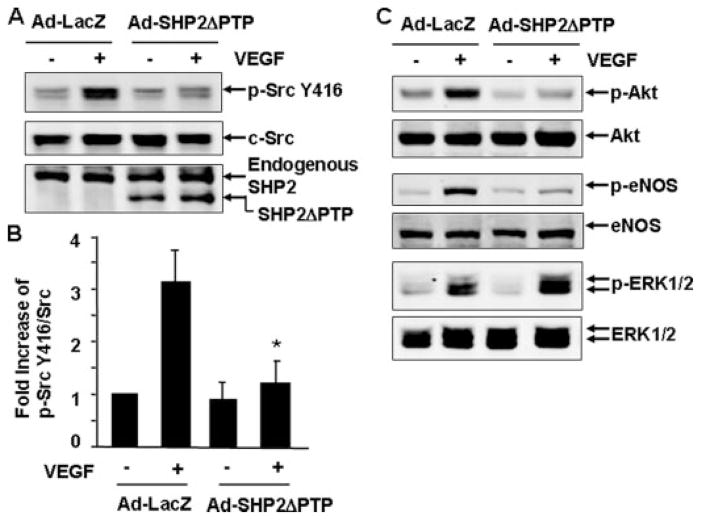
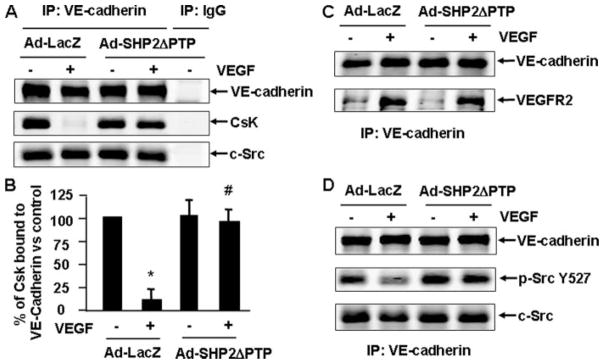
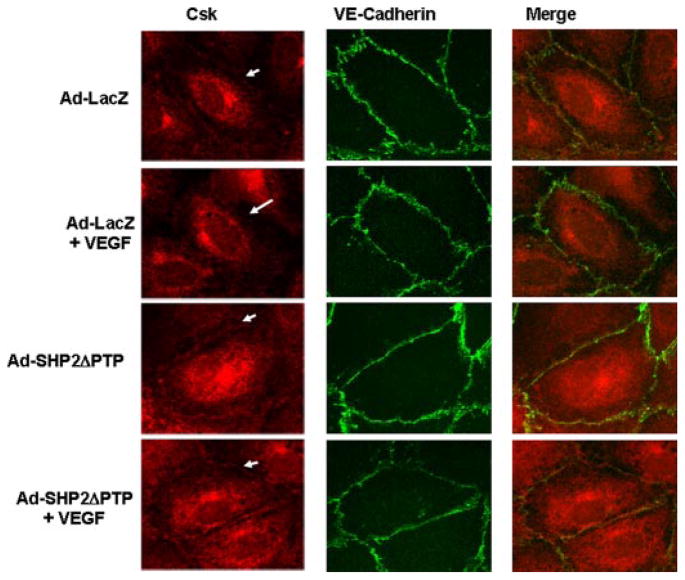
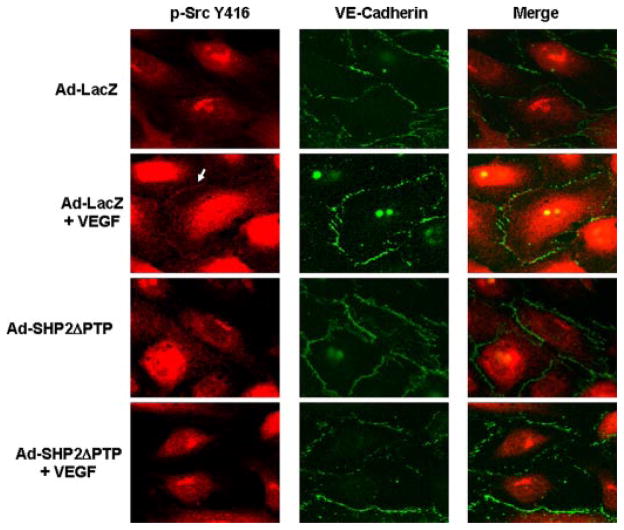
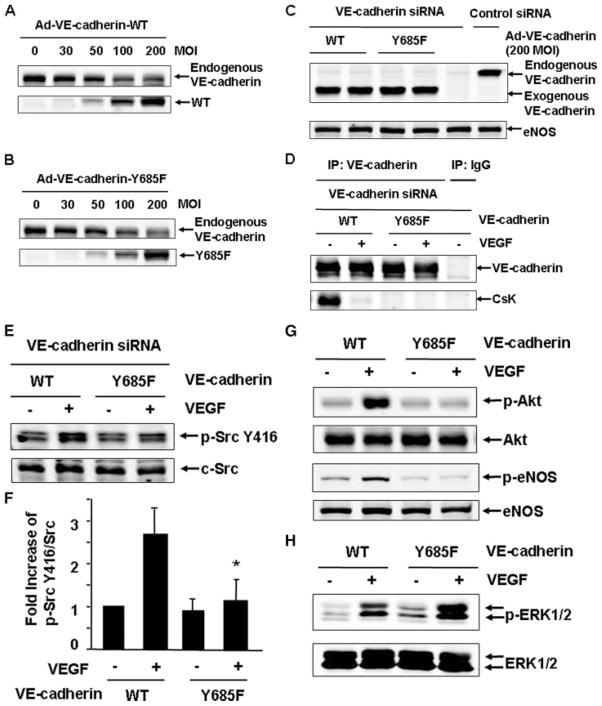
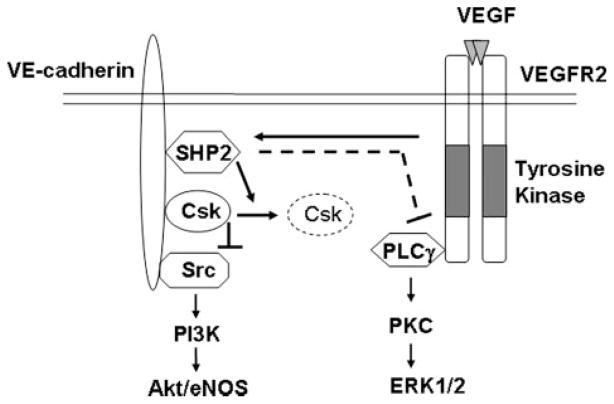
Vascular endothelial growth factor (VEGF) is a potent mediator of angiogenesis and vascular permeability, in which c-Src tyrosine kinase plays an essential role. However, the mechanisms by which VEGF stimulates c-Src activation have remained unclear. Here, we demonstrate that vascular endothelial cadherin (VE-cadherin) plays a critical role in regulating c-Src activation in response to VEGF. In vascular endothelial cells, VE-cadherin was basally associated with c-Src and Csk (C-terminal Src kinase), a negative regulator of Src activation. VEGF stimulated Csk release from VE-cadherin by recruiting the protein tyrosine phosphatase SHP2 to VE-cadherin signaling complex, leading to an increase in c-Src activation. Silencing VE-cadherin with small interference RNA significantly reduced VEGF-stimulated c-Src activation. Disrupting the association of VE-cadherin and Csk through the reconstitution of Csk binding-defective mutant of VE-cadherin also diminished Src activation. Moreover, inhibiting SHP2 by small interference RNA and adenovirus-mediated expression of a catalytically inactive mutant of SHP2 attenuated c-Src activation by blocking the disassociation of Csk from VE-cadherin. Furthermore, VE-cadherin and SHP2 differentially regulates VEGF downstream signaling. The inhibition of c-Src, VE-cadherin, and SHP2 diminished VEGF-mediated activation of Akt and endothelial nitric-oxide synthase. In contrast, inhibiting VE-cadherin and SHP2 enhanced ERK1/2 activation in response to VEGF. These findings reveal a novel role for VE-cadherin in modulating c-Src activation in VEGF signaling, thus providing new insights into the importance of VE-cadherin in VEGF signaling and vascular function.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
