

Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway
- PMID: 18180363
- PMCID: PMC2213608
- DOI: 10.1083/jcb.200710107
Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway
Abstract
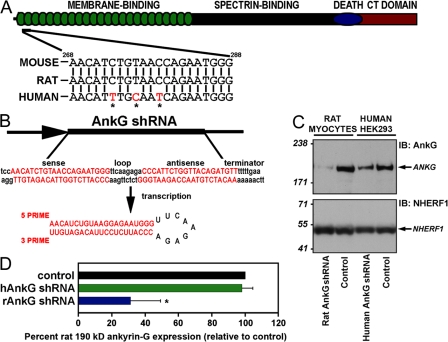
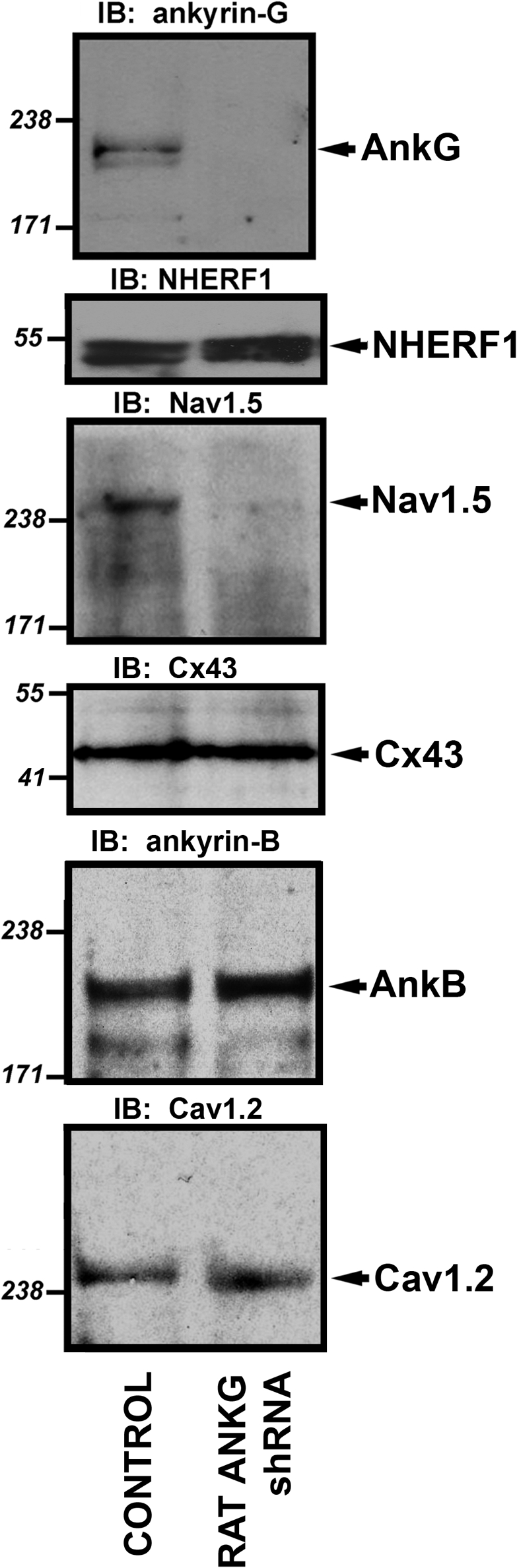
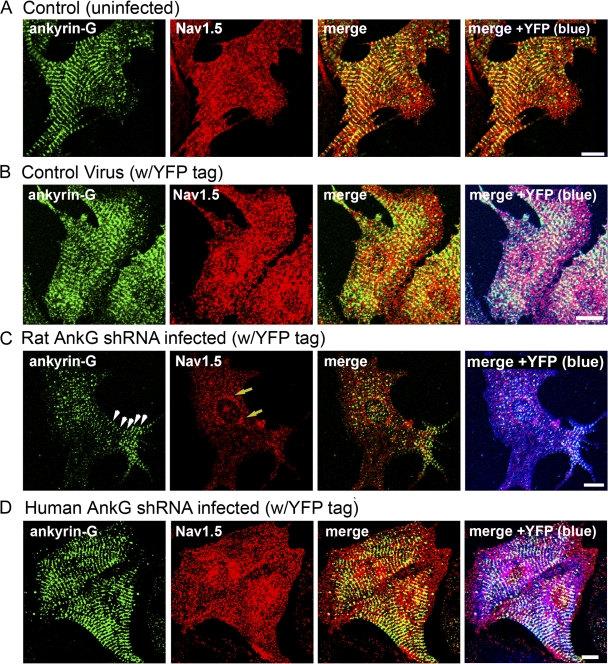
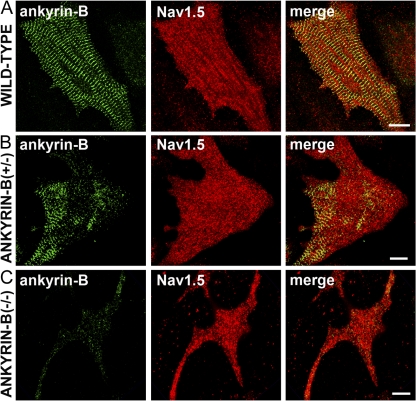
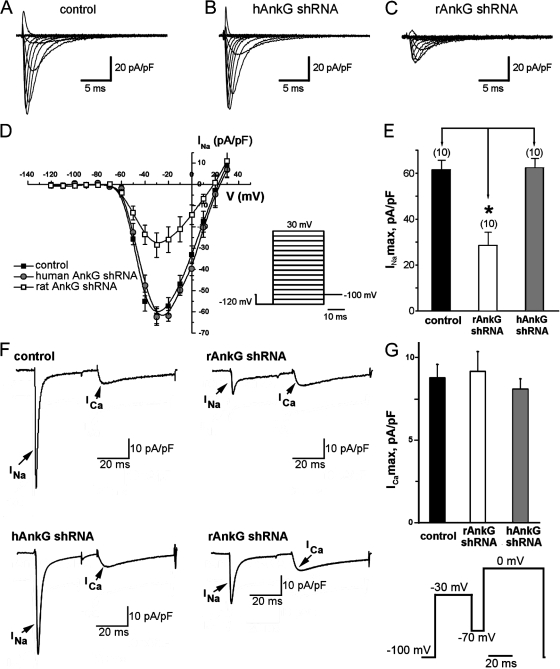
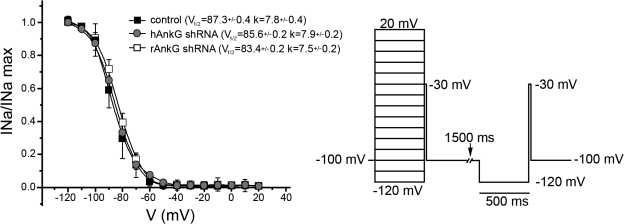
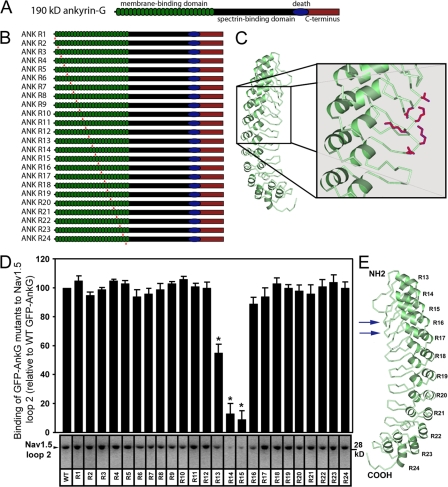
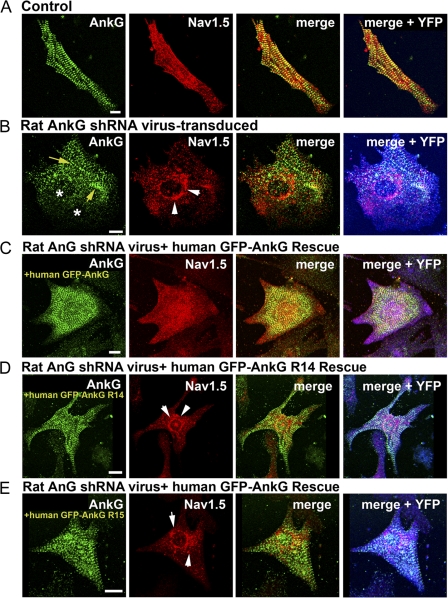
Voltage-gated Na(v) channels are required for normal electrical activity in neurons, skeletal muscle, and cardiomyocytes. In the heart, Na(v)1.5 is the predominant Na(v) channel, and Na(v)1.5-dependent activity regulates rapid upstroke of the cardiac action potential. Na(v)1.5 activity requires precise localization at specialized cardiomyocyte membrane domains. However, the molecular mechanisms underlying Na(v) channel trafficking in the heart are unknown. In this paper, we demonstrate that ankyrin-G is required for Na(v)1.5 targeting in the heart. Cardiomyocytes with reduced ankyrin-G display reduced Na(v)1.5 expression, abnormal Na(v)1.5 membrane targeting, and reduced Na(+) channel current density. We define the structural requirements on ankyrin-G for Na(v)1.5 interactions and demonstrate that loss of Na(v)1.5 targeting is caused by the loss of direct Na(v)1.5-ankyrin-G interaction. These data are the first report of a cellular pathway required for Na(v) channel trafficking in the heart and suggest that ankyrin-G is critical for cardiac depolarization and Na(v) channel organization in multiple excitable tissues.
Figures
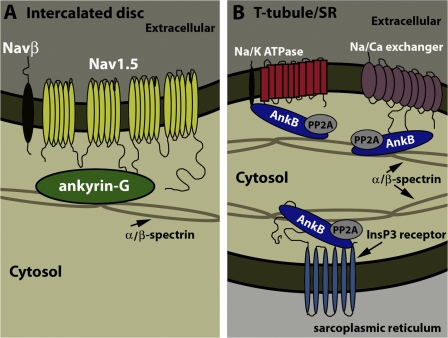
Comment in
-
Being there: cellular targeting of voltage-gated sodium channels in the heart.J Cell Biol. 2008 Jan 14;180(1):13-5. doi: 10.1083/jcb.200712098. Epub 2008 Jan 7. J Cell Biol. 2008. PMID: 18180365 Free PMC article.
References
-
- Abriel, H., and R.S. Kass. 2005. Regulation of the voltage-gated cardiac sodium channel Nav1.5 by interacting proteins. Trends Cardiovasc. Med. 15:35–40. - PubMed
-
- Anderson, C.L., B.P. Delisle, B.D. Anson, J.A. Kilby, M.L. Will, D.J. Tester, Q. Gong, Z. Zhou, M.J. Ackerman, and C.T. January. 2006. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 113:365–373. - PubMed
-
- Ballester, L.Y., D.W. Benson, B. Wong, I.H. Law, K.D. Mathews, C.G. Vanoye, and A.L. George Jr. 2006. Trafficking-competent and trafficking-defective KCNJ2 mutations in Andersen syndrome. Hum. Mutat. 27:388. - PubMed
-
- Barry, D.M., J.S. Trimmer, J.P. Merlie, and J.M. Nerbonne. 1995. Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ. Res. 77:361–369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
