

OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells
- PMID: 18182481
- PMCID: PMC2674576
- DOI: 10.1124/mol.107.042697
OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells
Abstract
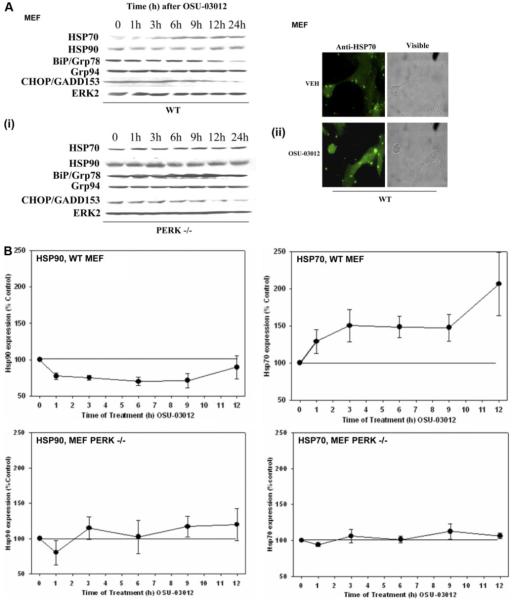
We have further defined mechanism(s) by which 2-amino-N-{4-[5-(2-phenanthrenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]-phenyl}acetamide [OSU-03012 (OSU)], a derivative of the cyclooxygenase-2 (COX2) inhibitor celecoxib but lacking COX2 inhibitory activity, kills transformed cells. In cells lacking expression of protein kinase R-like endoplasmic reticulum kinase (PERK(-/-)), the lethality of OSU was attenuated. OSU enhanced the expression of Beclin 1 and ATG5 and cleavage of pro-caspase 4 in a PERK-dependent fashion and promoted the Beclin 1- and ATG5-dependent formation of vacuoles containing LC3, followed by a subsequent caspase 4-dependent cleavage of cathepsin B and a cathepsin B-dependent formation of low pH intracellular vesicles; cathepsin B was activated and released into the cytosol and genetic suppression of caspase 4, cathepsin B, or apoptosis-inducing factor function significantly suppressed cell killing. In parallel, OSU caused PERK-dependent increases in 70-kDa heat shock protein (HSP70) expression and decreases in 90-kDa heat shock protein (HSP90) and Grp78/BiP expression. Changes in HSP70 expression were post-transcriptional. Knock-down or small-molecule inhibition of HSP70 expression enhanced OSU toxicity, and overexpression of HSP70 suppressed OSU-induced low pH vesicle formation and lethality. Our data demonstrate that OSU-03012 causes cell killing that is dependent on PERK-induced activation of multiple toxic proteases. OSU-03012 also increased expression of HSP70 in a PERK-dependent fashion, providing support for the contention that OSU-03012-induced PERK signaling promotes both cell survival and cell death processes.
Figures








References
-
- Carón RW, Yacoub A, Li M, Zhu X, Mitchell C, Hong Y, Hawkins W, Sasazuki T, Shirasawa S, Kozikowski AP, Dennis PA, Hagan MP, Grant S, Dent P. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Ther. 2005;4:257–270. - PubMed
-
- Cui W, Yu CH, Hu KQ. In vitro and in vivo effects and mechanisms of celecoxib-induced growth inhibition of human hepatocellular carcinoma cells. Clin Cancer Res. 2005;11:8213–8221. - PubMed
-
- Demidenko ZN, Vivo C, Halicka HD, Li CJ, Bhalla K, Broude EV, Blagoskonny MV. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: comparison with caspase inhibitor- and cycle-arrest-mediated cyto-protection. Cell Death Differ. 2006;13:1434–1441. - PubMed
-
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. - PubMed
-
- Dunn WA. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994;4:139–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
