

Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation
- PMID: 18184782
- PMCID: PMC6670512
- DOI: 10.1523/JNEUROSCI.3796-07.2008
Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation
Abstract
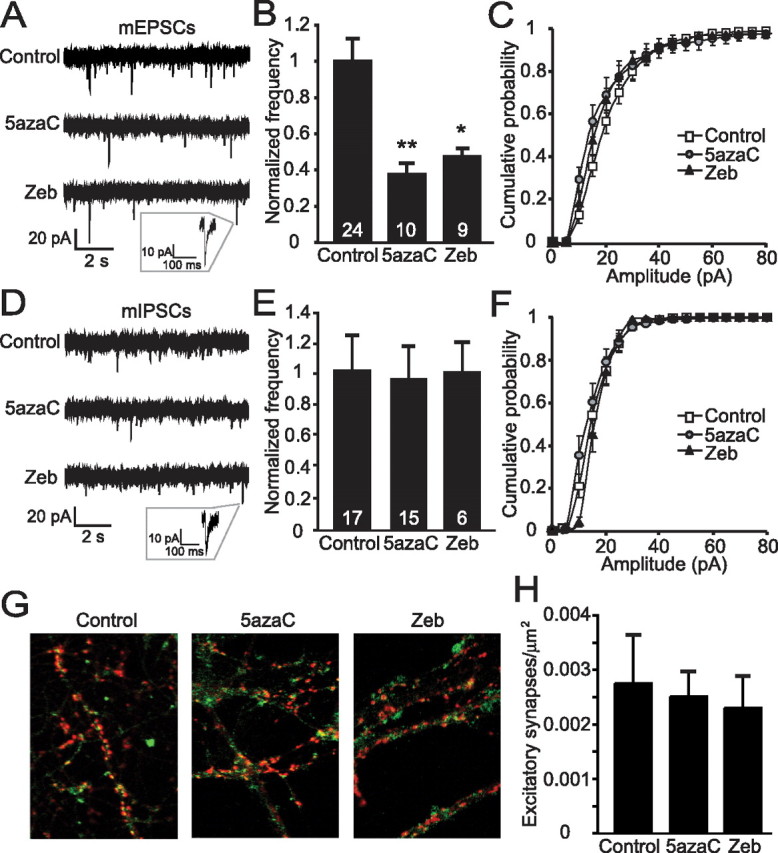
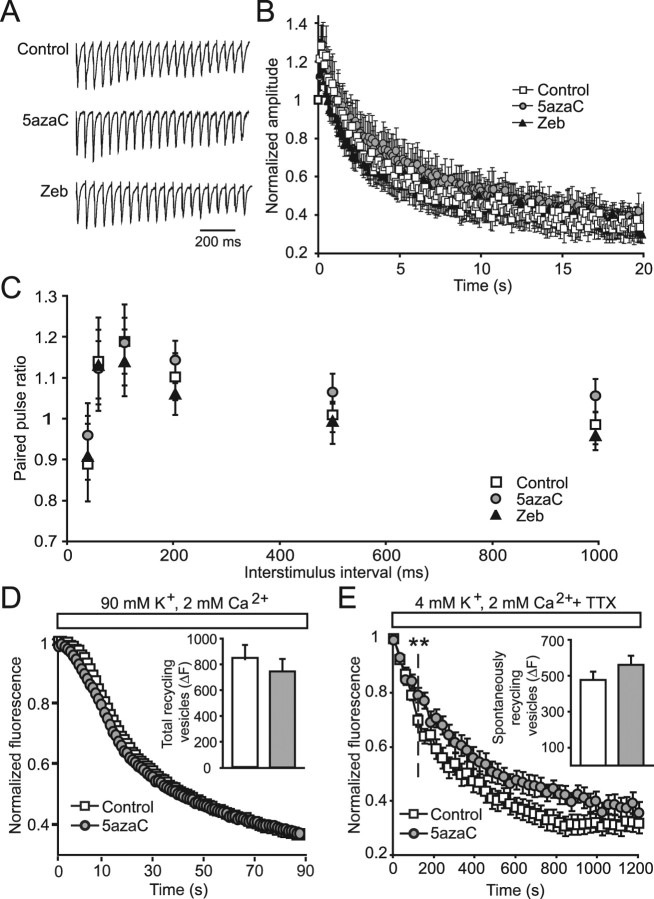
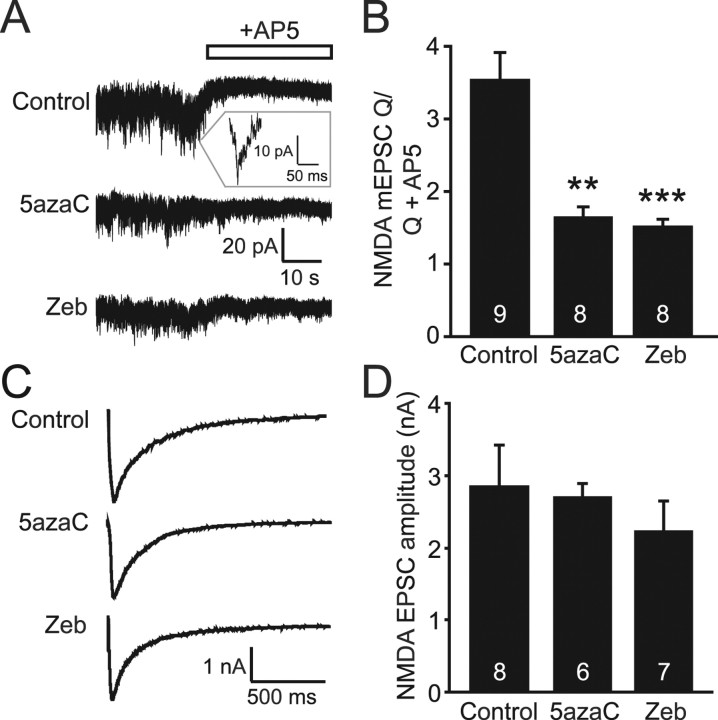
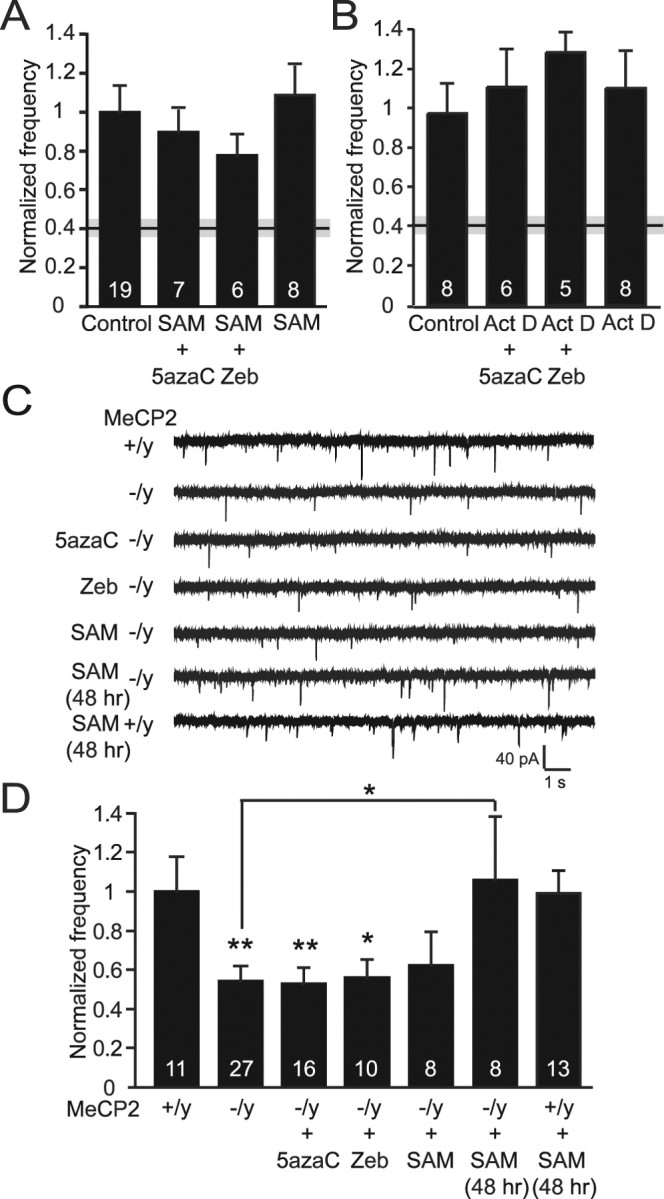
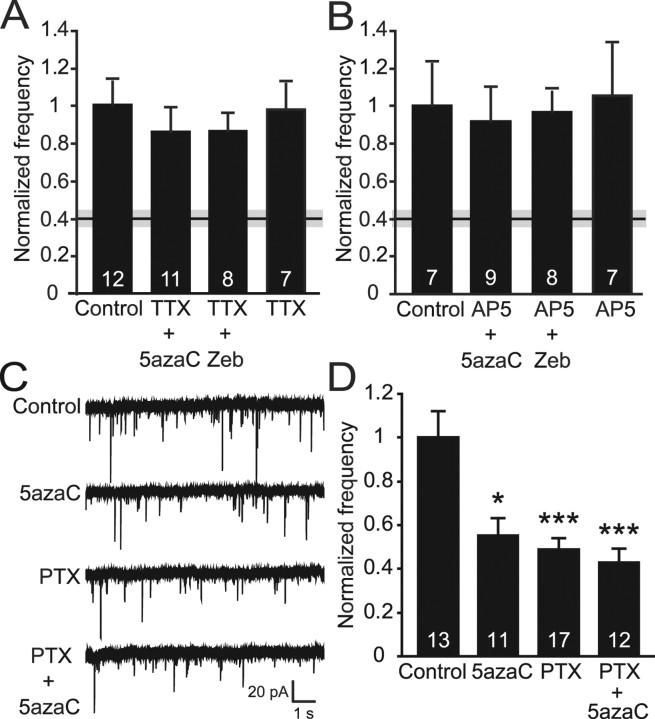
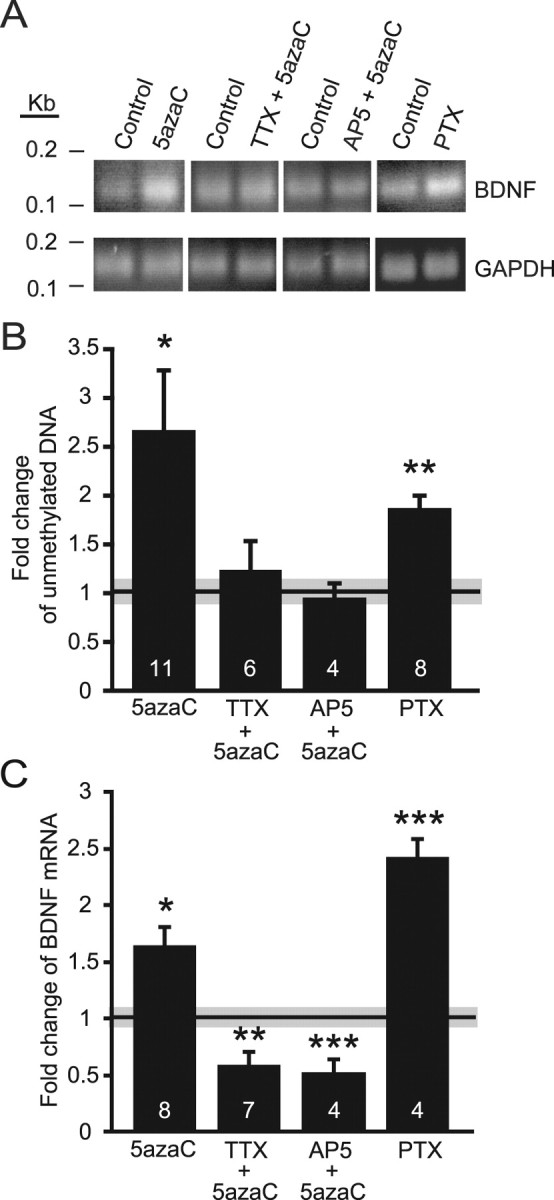
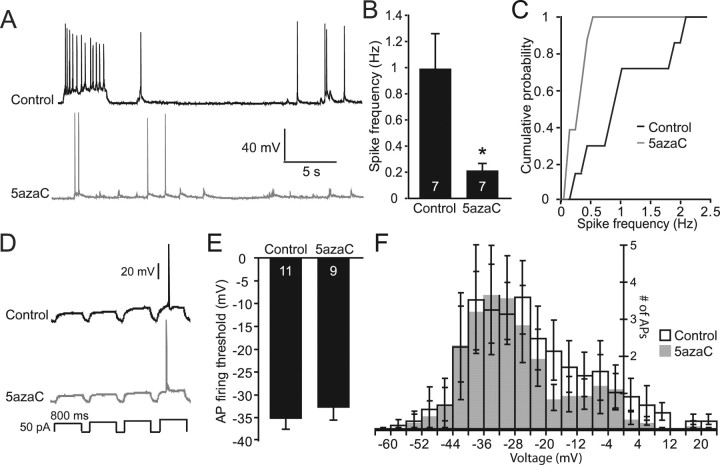
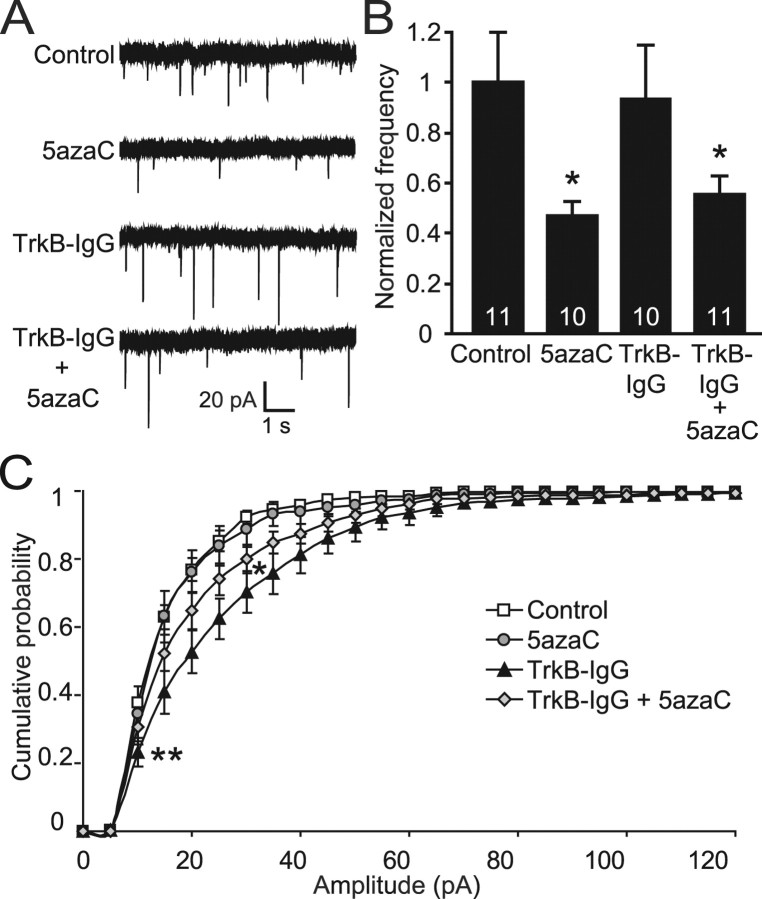
DNA methylation is an epigenetic mechanism that plays a critical role in the repression of gene expression. Here, we show that DNA methyltransferase (DNMT) inhibition in hippocampal neurons results in activity-dependent demethylation of genomic DNA and a parallel decrease in the frequency of miniature EPSCs (mEPSCs), which in turn impacts neuronal excitability and network activity. Treatment with DNMT inhibitors reveals an activity-driven demethylation of brain-derived neurotrophic factor promoter I, which is mediated by synaptic activation of NMDA receptors, because it is susceptible to AP-5, a blocker of NMDA receptors. The specific effect of DNMT inhibition on spontaneous excitatory neurotransmission requires gene transcription and is occluded in the absence of the transcriptional repressor methyl-CpG-binding protein 2 (MeCP2). Interestingly, enhancing excitatory activity, in the absence of DNMT inhibitors, also produces similar decreases in DNA methylation and mEPSC frequency, suggesting a role for DNA methylation in the control of homeostatic synaptic plasticity. Furthermore, adding excess substrate for DNA methylation (S-adenosyl-L-methionine) rescues the suppression of mEPSCs by DNMT inhibitors in wild-type neurons, as well as the defect seen in MeCP2-deficient neurons. These results uncover a means by which NMDA receptor-mediated synaptic activity drives DNA demethylation within mature neurons and suppresses basal synaptic function.
Figures
References
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature. 2007;445:671–675. - PubMed
-
- Betz WJ, Mao F, Smith CB. Imaging exocytosis and endocytosis. Curr Opin Neurobiol. 1996;6:365–371. - PubMed
-
- Bito H, Deisseroth K, Tsien RW. Ca2+-dependent regulation in neuronal gene expression. Curr Opin Neurobiol. 1997;7:419–429. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources