

Universality and diversity of folding mechanics for three-helix bundle proteins
- PMID: 18195374
- PMCID: PMC2242684
- DOI: 10.1073/pnas.0707284105
Universality and diversity of folding mechanics for three-helix bundle proteins
Abstract
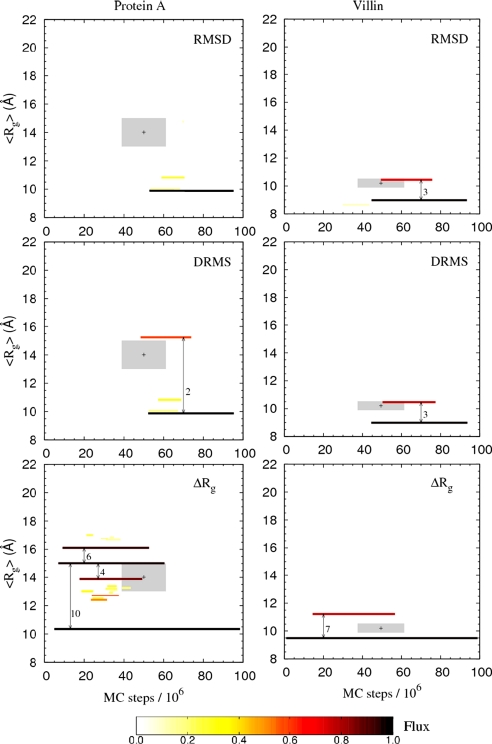
In this study we evaluate, at full atomic detail, the folding processes of two small helical proteins, the B domain of protein A and the Villin headpiece. Folding kinetics are studied by performing a large number of ab initio Monte Carlo folding simulations using a single transferable all-atom potential. Using these trajectories, we examine the relaxation behavior, secondary structure formation, and transition-state ensembles (TSEs) of the two proteins and compare our results with experimental data and previous computational studies. To obtain a detailed structural information on the folding dynamics viewed as an ensemble process, we perform a clustering analysis procedure based on graph theory. Moreover, rigorous p(fold) analysis is used to obtain representative samples of the TSEs and a good quantitative agreement between experimental and simulated Phi values is obtained for protein A. Phi values for Villin also are obtained and left as predictions to be tested by future experiments. Our analysis shows that the two-helix hairpin is a common partially stable structural motif that gets formed before entering the TSE in the studied proteins. These results together with our earlier study of Engrailed Homeodomain and recent experimental studies provide a comprehensive, atomic-level picture of folding mechanics of three-helix bundle proteins.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Alm E, Baker D. Curr Opin Struct Biol. 1999;9:189–196. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
